

Non-Glycanated Biglycan and LTBP4: Leveraging the extracellular matrix for Duchenne Muscular Dystrophy therapeutics
- PMID: 29481844
- PMCID: PMC6015525
- DOI: 10.1016/j.matbio.2018.02.016
Non-Glycanated Biglycan and LTBP4: Leveraging the extracellular matrix for Duchenne Muscular Dystrophy therapeutics
Abstract
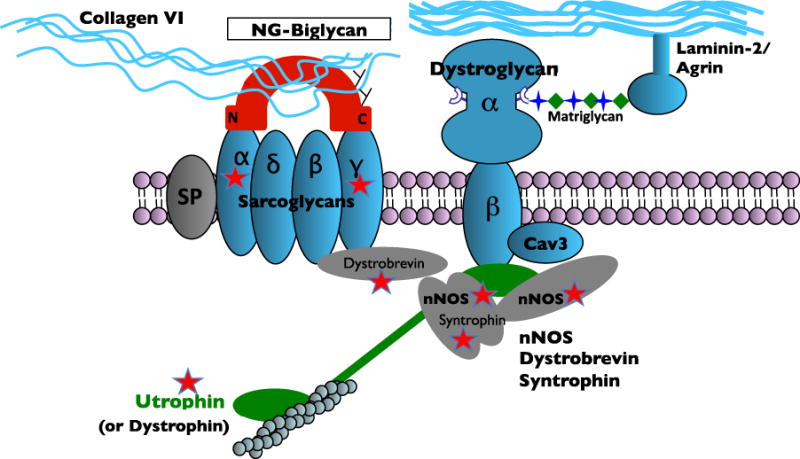
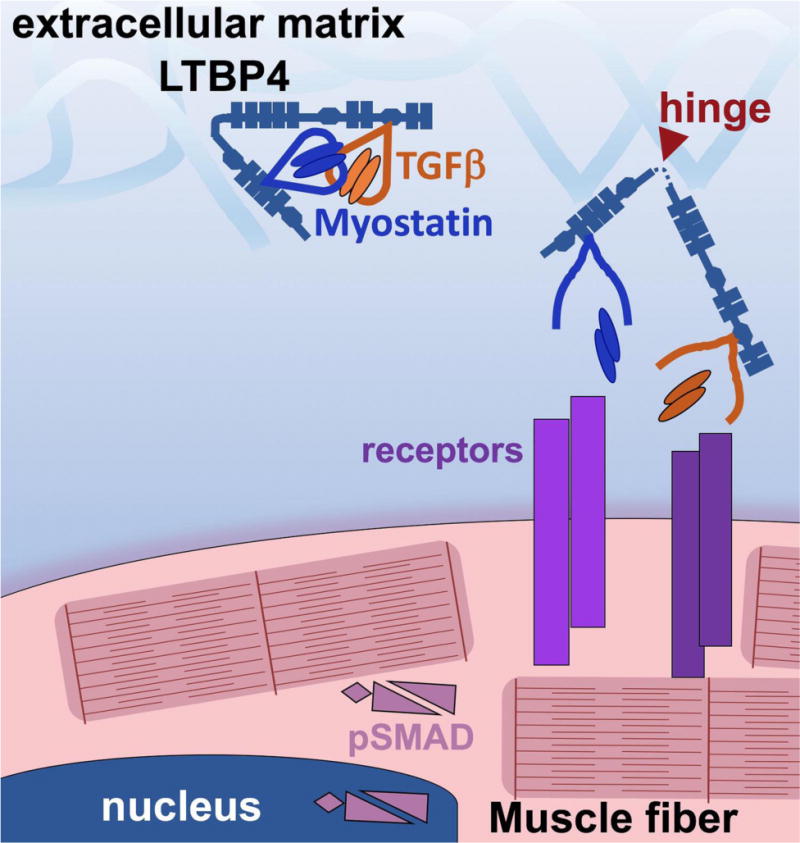
The extracellular matrix (ECM) plays key roles in normal and diseased skeletal and cardiac muscle. In healthy muscle the ECM is essential for transmitting contractile force, maintaining myofiber integrity and orchestrating cellular signaling. Duchenne Muscular Dystrophy (DMD) is caused by loss of dystrophin, a cytosolic protein that anchors a transmembrane complex and serves as a vital link between the actin cytoskeleton and the basal lamina. Loss of dystrophin leads to membrane fragility and impaired signaling, resulting in myofiber death and cycles of inflammation and regeneration. Fibrosis is also a cardinal feature of DMD. In this review, we will focus on two cases where understanding the normal function and regulation of ECM in muscle has led to the discovery of candidate therapeutics for DMD. Biglycan is a small leucine rich repeat ECM protein present as two glycoforms in muscle that have dramatically different functions. One widely expressed form is biglycan proteoglycan (PG) that bears two chondroitin sulfate GAG chains (typically chondroitin sulfate) and two N-linked carbohydrates. The second glycoform, referred to as 'NG' (non-glycanated) biglycan, lacks the GAG side chains. NG, but not PG biglycan recruits utrophin, an autosomal paralog of dystrophin, and an NOS-containing signaling complex to the muscle cell membrane. Recombinant NG biglycan can be systemically delivered to dystrophic mice where it upregulates utrophin at the membrane and improves muscle health and function. An optimized version of NG biglycan, 'TVN-102', is under development as a candidate therapeutic for DMD. A second matrix-embedded protein being evaluated for therapeutic potential is latent TGFβ binding protein 4 (LTBP4). Identified in a genomic screen for modifiers of muscular dystrophy, LTBP4 binds both TGFβ and myostatin. Genetic studies identified the hinge region of LTBP4 as linked to TGFβ release and contributing to the "hyper-TGFβ" signaling state that promotes fibrosis in muscular dystrophy. This hinge region can be stabilized by antibodies directed towards this domain. Stabilizing the hinge region of LTBP4 is expected to reduce latent TGFβ release and thus reduce fibrosis.
Copyright © 2018 Elsevier B.V. All rights reserved.
Figures

References
-
- Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, Sampson JB, Mendell JR, Wall C, King WM, Pestronk A, Florence JM, Connolly AM, Mathews KD, Stephan CM, Laubenthal KS, Wong BL, Morehart PJ, Meyer A, Finkel RS, Bonnemann CG, Medne L, Day JW, Dalton JC, Margolis MK, Hinton VJ, C. United Dystrophinopathy Project. Weiss RB. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Human mutation. 2009;30(12):1657–66. - PMC - PubMed
-
- Kirschner J, Lochmuller H. Sarcoglycanopathies. Handbook of clinical neurology. 2011;101:41–6. - PubMed
-
- Laval SH, Bushby KM. Limb-girdle muscular dystrophies–from genetics to molecular pathology. Neuropathology and applied neurobiology. 2004;30(2):91–105. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
