

Efficacy of Exome-Targeted Capture Sequencing to Detect Mutations in Known Cerebellar Ataxia Genes
- PMID: 29482223
- PMCID: PMC5885259
- DOI: 10.1001/jamaneurol.2017.5121
Efficacy of Exome-Targeted Capture Sequencing to Detect Mutations in Known Cerebellar Ataxia Genes
Abstract
Importance: Molecular diagnosis is difficult to achieve in disease groups with a highly heterogeneous genetic background, such as cerebellar ataxia (CA). In many patients, candidate gene sequencing or focused resequencing arrays do not allow investigators to reach a genetic conclusion.
Objectives: To assess the efficacy of exome-targeted capture sequencing to detect mutations in genes broadly linked to CA in a large cohort of undiagnosed patients and to investigate their prevalence.
Design, setting, and participants: Three hundred nineteen index patients with CA and without a history of dominant transmission were included in the this cohort study by the Spastic Paraplegia and Ataxia Network. Centralized storage was in the DNA and cell bank of the Brain and Spine Institute, Salpetriere Hospital, Paris, France. Patients were classified into 6 clinical groups, with the largest being those with spastic ataxia (ie, CA with pyramidal signs [n = 100]). Sequencing was performed from January 1, 2014, through December 31, 2016. Detected variants were classified as very probably or definitely causative, possibly causative, or of unknown significance based on genetic evidence and genotype-phenotype considerations.
Main outcomes and measures: Identification of variants in genes broadly linked to CA, classified in pathogenicity groups.
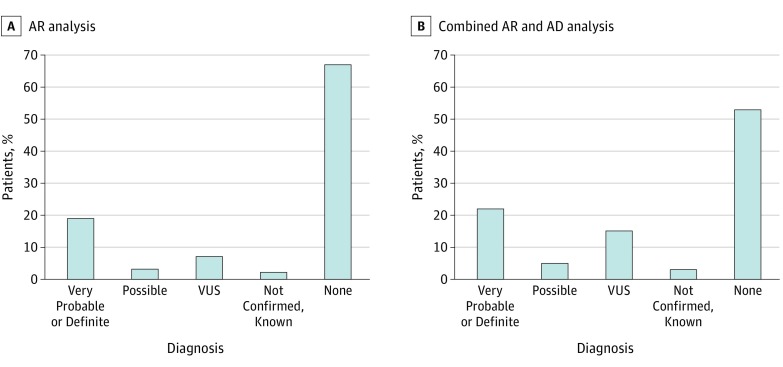
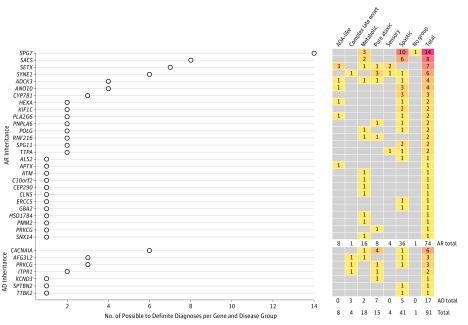
Results: The 319 included patients had equal sex distribution (160 female [50.2%] and 159 male patients [49.8%]; mean [SD] age at onset, 27.9 [18.6] years). The age at onset was younger than 25 years for 131 of 298 patients (44.0%) with complete clinical information. Consanguinity was present in 101 of 298 (33.9%). Very probable or definite diagnoses were achieved for 72 patients (22.6%), with an additional 19 (6.0%) harboring possibly pathogenic variants. The most frequently mutated genes were SPG7 (n = 14), SACS (n = 8), SETX (n = 7), SYNE1 (n = 6), and CACNA1A (n = 6). The highest diagnostic rate was obtained for patients with an autosomal recessive CA with oculomotor apraxia-like phenotype (6 of 17 [35.3%]) or spastic ataxia (35 of 100 [35.0%]) and patients with onset before 25 years of age (41 of 131 [31.3%]). Peculiar phenotypes were reported for patients carrying KCND3 or ERCC5 variants.
Conclusions and relevance: Exome capture followed by targeted analysis allows the molecular diagnosis in patients with highly heterogeneous mendelian disorders, such as CA, without prior assumption of the inheritance mode or causative gene. Being commonly available without specific design need, this procedure allows testing of a broader range of genes, consequently describing less classic phenotype-genotype correlations, and post hoc reanalysis of data as new genes are implicated in the disease.
Conflict of interest statement
Figures
References
-
- Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N Engl J Med. 2012;366(7):636-646. - PubMed
-
- Campuzano V, Montermini L, Lutz Y, et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet. 1997;6(11):1771-1780. - PubMed
-
- Savitsky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268(5218):1749-1753. - PubMed
-
- Lecocq C, Charles P, Azulay JP, et al. Delayed-onset Friedreich’s ataxia revisited. Mov Disord. 2016;31(1):62-69. - PubMed
-
- Méneret A, Ahmar-Beaugendre Y, Rieunier G, et al. The pleiotropic movement disorders phenotype of adult ataxia-telangiectasia. Neurology. 2014;83(12):1087-1095. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
