

Hypoxia-inducible factor 2-alpha-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury
- PMID: 29483579
- PMCID: PMC5827027
- DOI: 10.1038/s41467-018-03105-2
Hypoxia-inducible factor 2-alpha-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury
Abstract
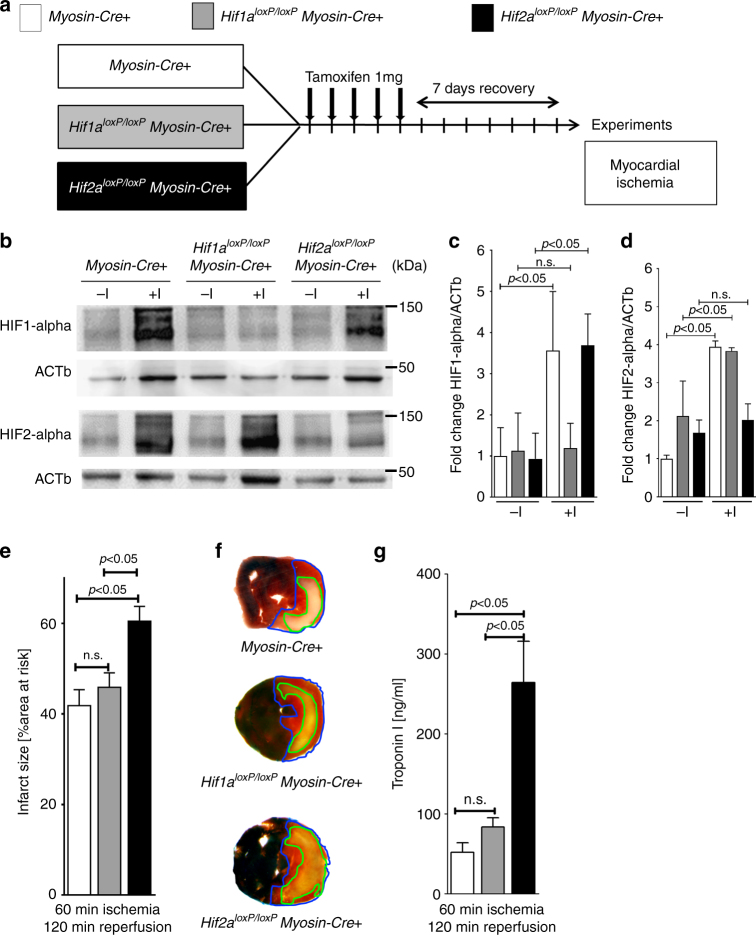
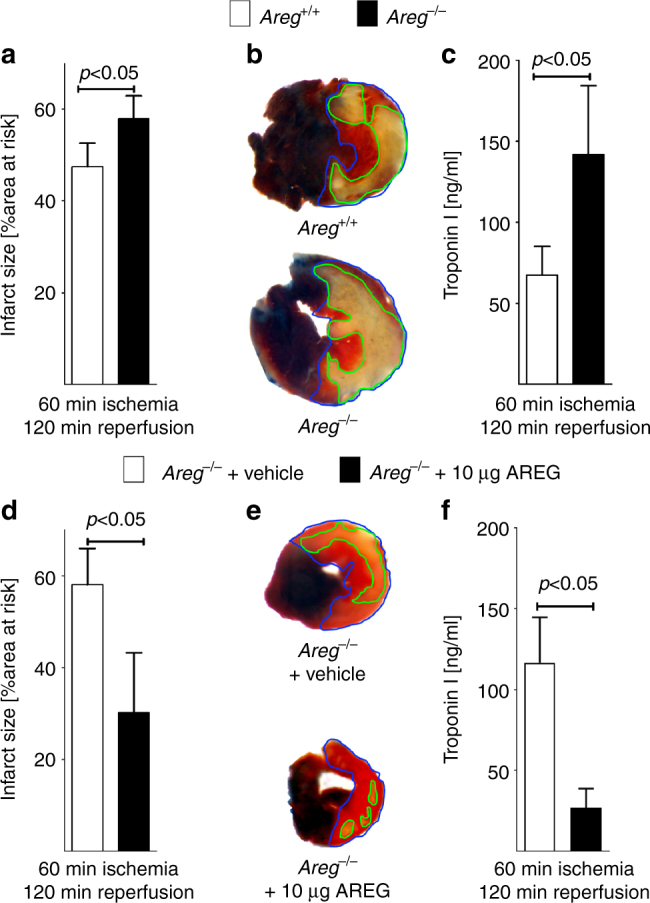
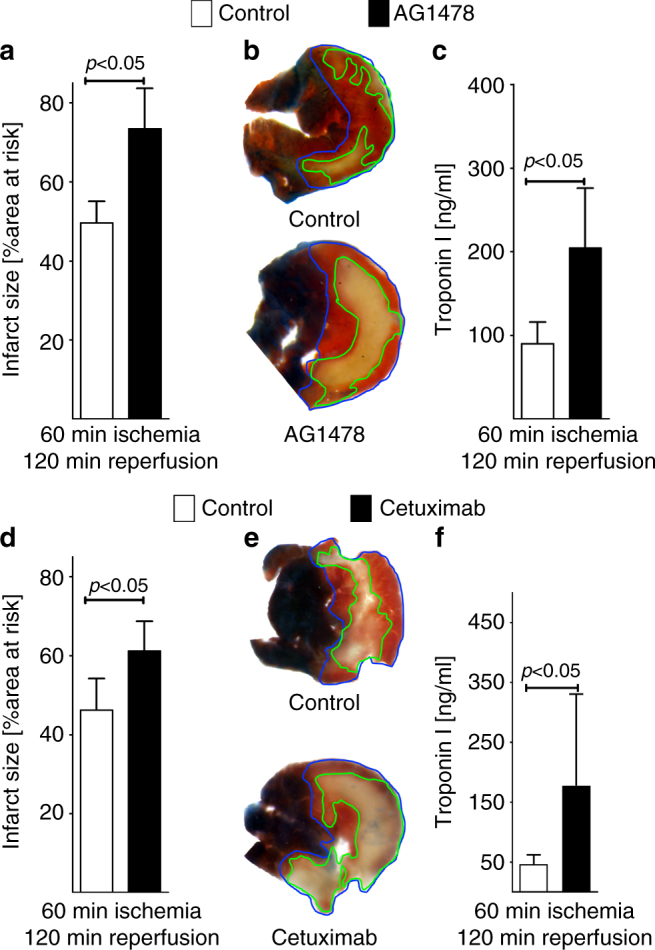
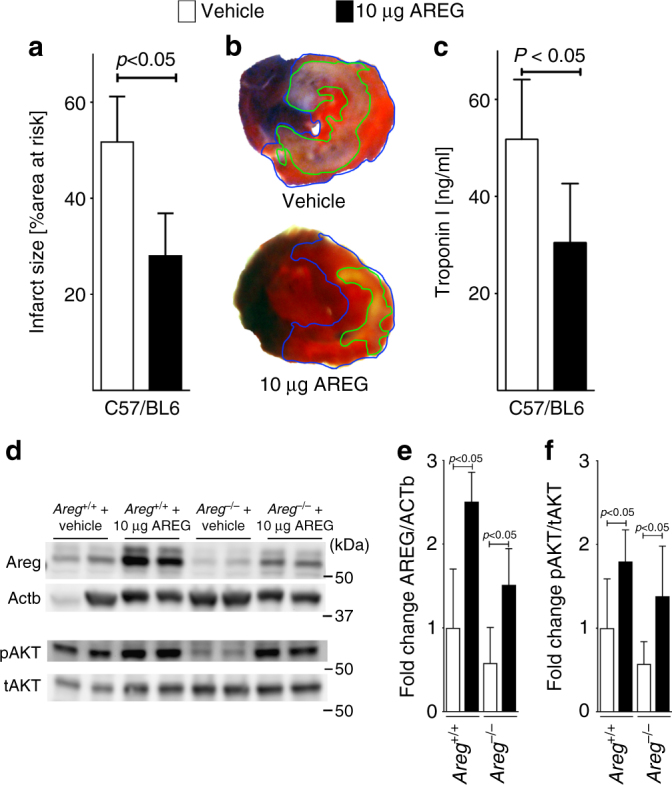
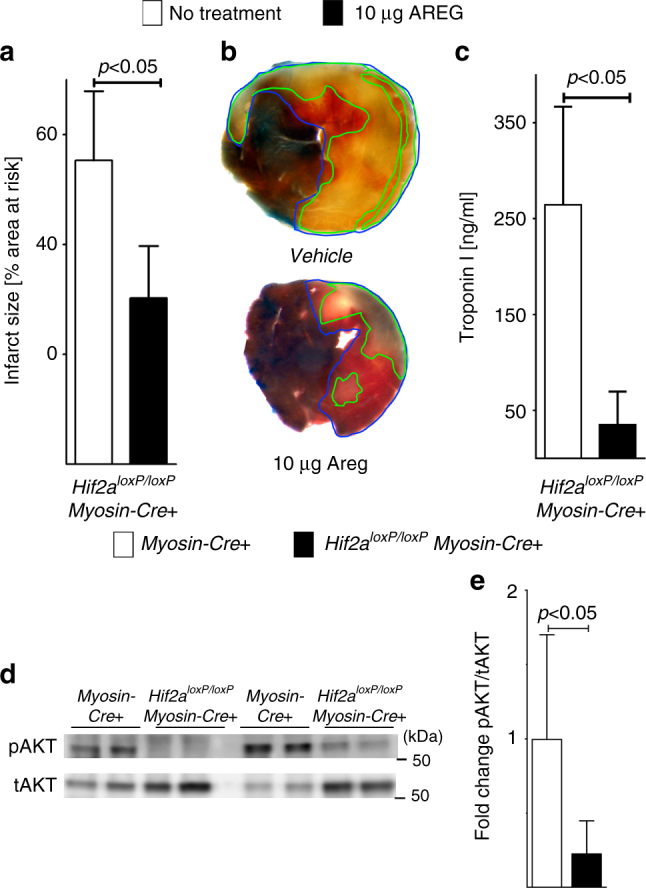
Myocardial ischemia-reperfusion injury (IRI) leads to the stabilization of the transcription factors hypoxia-inducible factor 1-alpha (HIF1-alpha) and hypoxia-inducible factor 2-alpha (HIF2-alpha). While previous studies implicate HIF1-alpha in cardioprotection, the role of HIF2-alpha remains elusive. Here we show that HIF2-alpha induces the epithelial growth factor amphiregulin (AREG) to elicit cardioprotection in myocardial IRI. Comparing mice with inducible deletion of Hif1a or Hif2a in cardiac myocytes, we show that loss of Hif2-alpha increases infarct sizes. Microarray studies in genetic models or cultured human cardiac myocytes implicate HIF2-alpha in the myocardial induction of AREG. Likewise, AREG increases in myocardial tissues from patients with ischemic heart disease. Areg deficiency increases myocardial IRI, as does pharmacologic inhibition of Areg signaling. In contrast, treatment with recombinant Areg provides cardioprotection and reconstitutes mice with Hif2a deletion. These studies indicate that HIF2-alpha induces myocardial AREG expression in cardiac myocytes, which increases myocardial ischemia tolerance.
Conflict of interest statement
The authors declare no competing financial interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
