

Accounting for Errors in Low Coverage High-Throughput Sequencing Data When Constructing Genetic Maps Using Biparental Outcrossed Populations
- PMID: 29487138
- PMCID: PMC5937187
- DOI: 10.1534/genetics.117.300627
Accounting for Errors in Low Coverage High-Throughput Sequencing Data When Constructing Genetic Maps Using Biparental Outcrossed Populations
Abstract
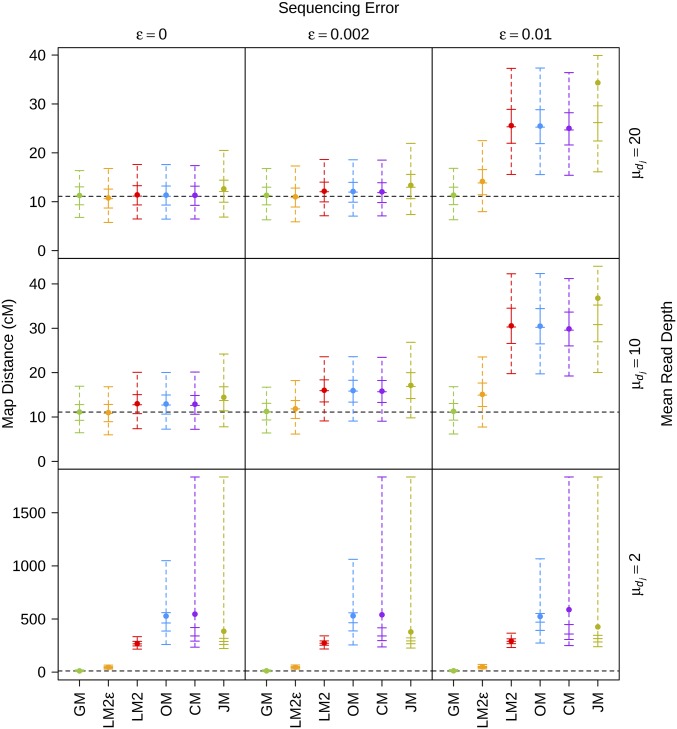
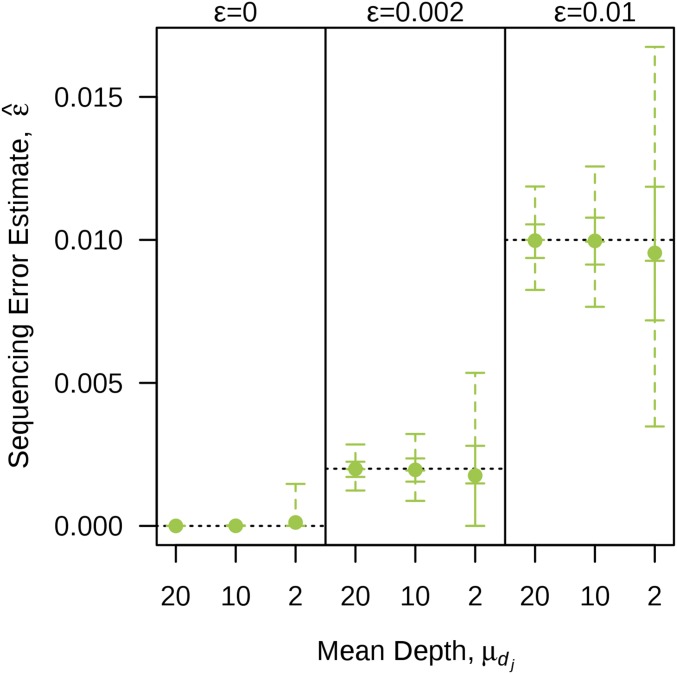
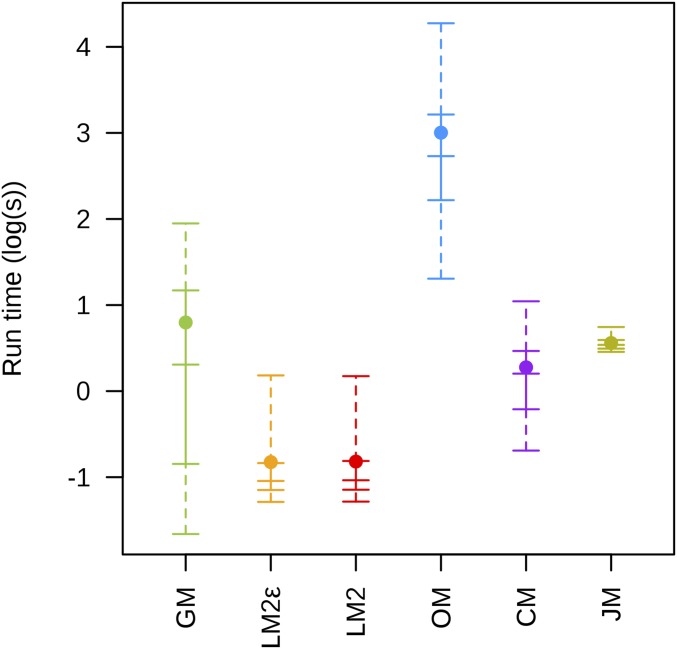
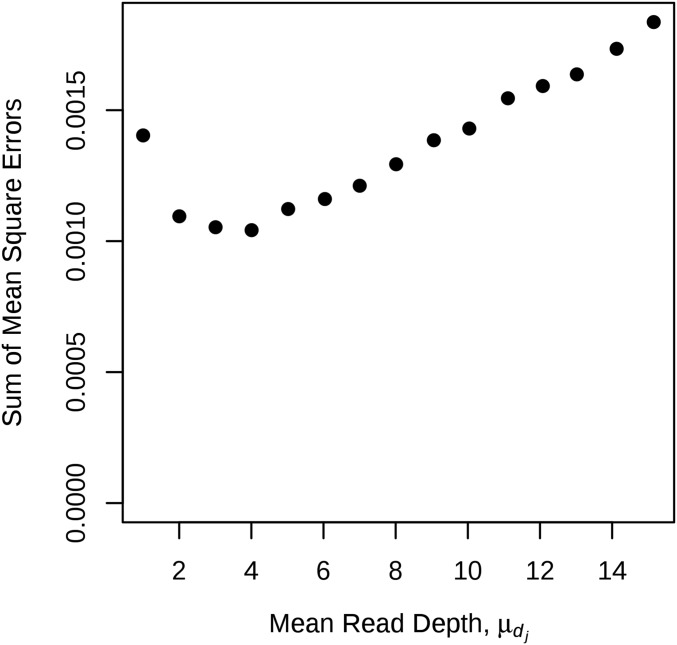
Next-generation sequencing is an efficient method that allows for substantially more markers than previous technologies, providing opportunities for building high-density genetic linkage maps, which facilitate the development of nonmodel species' genomic assemblies and the investigation of their genes. However, constructing genetic maps using data generated via high-throughput sequencing technology (e.g., genotyping-by-sequencing) is complicated by the presence of sequencing errors and genotyping errors resulting from missing parental alleles due to low sequencing depth. If unaccounted for, these errors lead to inflated genetic maps. In addition, map construction in many species is performed using full-sibling family populations derived from the outcrossing of two individuals, where unknown parental phase and varying segregation types further complicate construction. We present a new methodology for modeling low coverage sequencing data in the construction of genetic linkage maps using full-sibling populations of diploid species, implemented in a package called GUSMap. Our model is based on the Lander-Green hidden Markov model but extended to account for errors present in sequencing data. We were able to obtain accurate estimates of the recombination fractions and overall map distance using GUSMap, while most existing mapping packages produced inflated genetic maps in the presence of errors. Our results demonstrate the feasibility of using low coverage sequencing data to produce genetic maps without requiring extensive filtering of potentially erroneous genotypes, provided that the associated errors are correctly accounted for in the model.
Keywords: genetic linkage maps; genotyping-by-sequencing; hidden Markov model; map inflation; sequencing errors.
Copyright © 2018 Bilton et al.
Figures
References
-
- Baum L. E., Petrie T., Soules G., Weiss N., 1970. A maximization technique occurring in the statistical analysis of probabilistic functions of Markov chains. Ann. Math. Stat. 41: 164–171. 10.1214/aoms/1177697196 - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
