

Managing Bardet-Biedl Syndrome-Now and in the Future
- PMID: 29487844
- PMCID: PMC5816783
- DOI: 10.3389/fped.2018.00023
Managing Bardet-Biedl Syndrome-Now and in the Future
Abstract
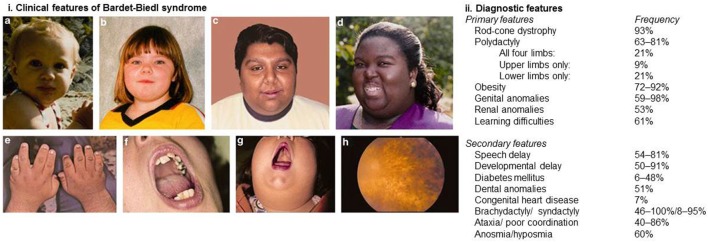
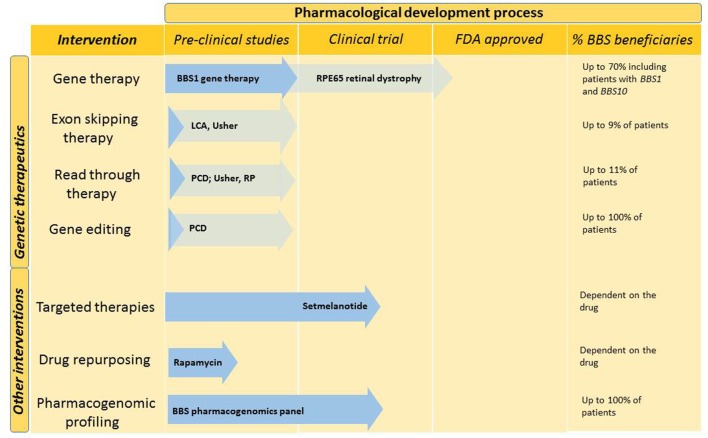
Bardet-Biedl syndrome is a rare autosomal recessive multisystem disorder caused by defects in genes encoding for proteins that localize to the primary cilium/basal body complex. Twenty-one disease-causing genes have been identified to date. It is one of the most well-studied conditions in the family of diseases caused by defective cilia collectively known as ciliopathies. In this review, we provide an update on diagnostic developments, clinical features, and progress in the management of Bardet-Biedl syndrome. Advances in diagnostic technologies including exome and whole genome sequencing are expanding the spectrum of patients who are diagnosed with Bardet-Biedl syndrome and increasing the number of cases with diagnostic uncertainty. As a result of the diagnostic developments, a small number of patients with only one or two clinical features of Bardet-Biedl syndrome are being diagnosed. Our understanding of the syndrome-associated renal disease has evolved and is reviewed here. Novel interventions are developing at a rapid pace and are explored in this review including genetic therapeutics such as gene therapy, exon skipping therapy, nonsense suppression therapy, and gene editing. Other non-genetic therapies such as gene repurposing, targeted therapies, and non-pharmacological interventions are also discussed.
Keywords: Bardet–Biedl syndrome; drug repurposing; genetic therapies; genome editing; pharmacogenomics; targeted therapies.
Figures
References
-
- Forsythe E, Beales PL. Bardet-Biedl syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews(R). Seattle, WA: (2003).
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials