

The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency
- PMID: 29489750
- PMCID: PMC5951268
- DOI: 10.1038/nature25784
The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency
Abstract
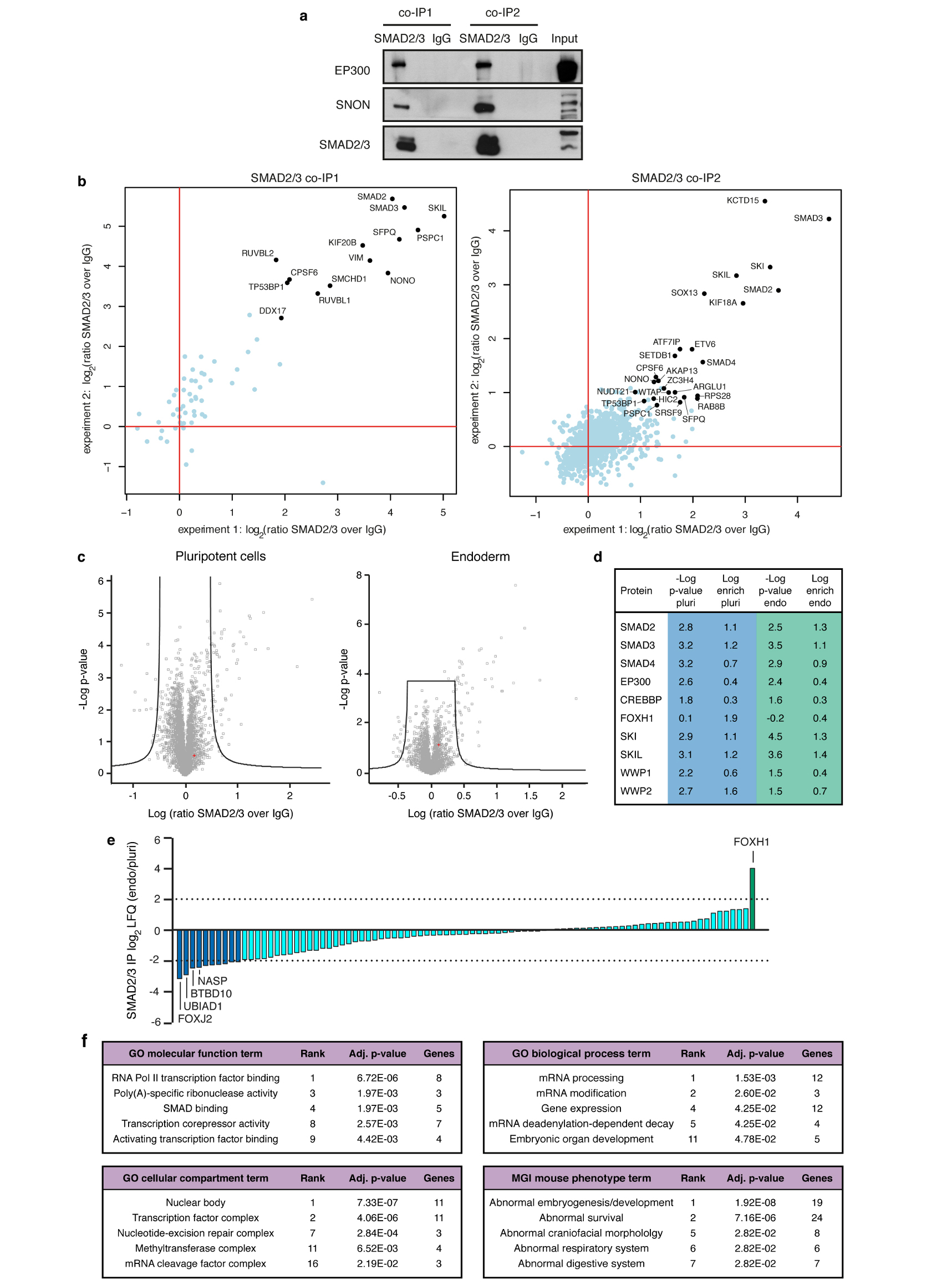
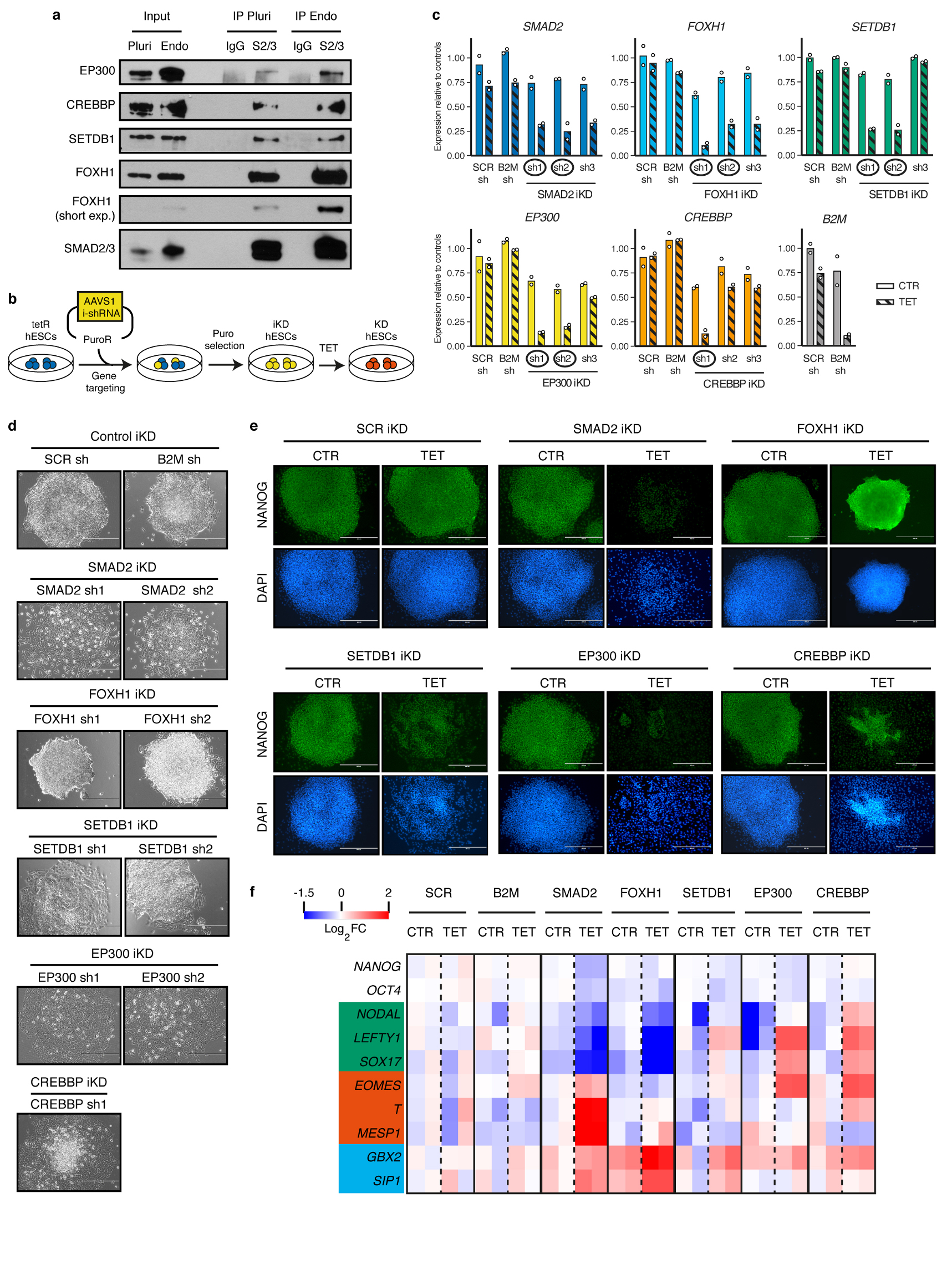
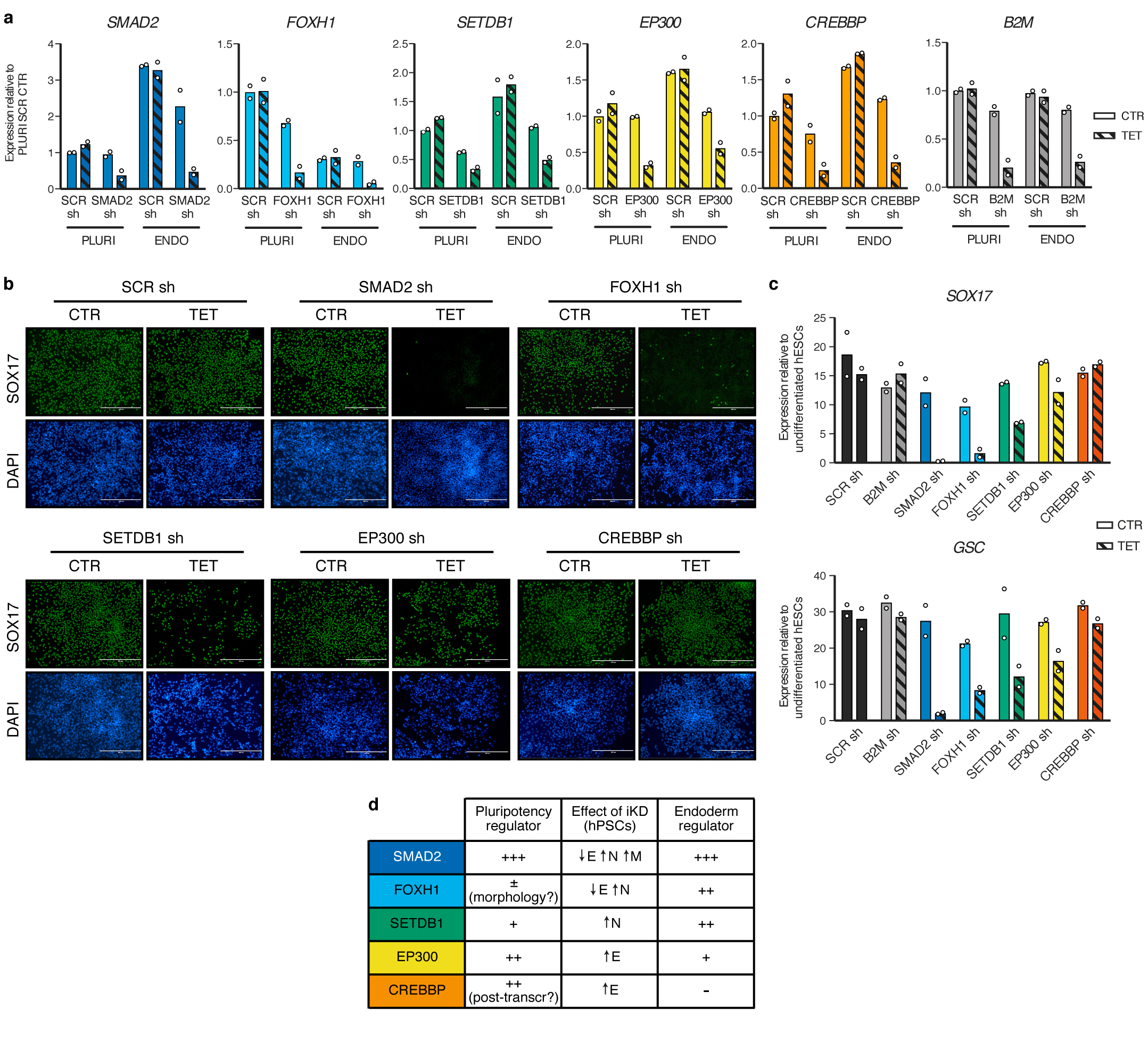
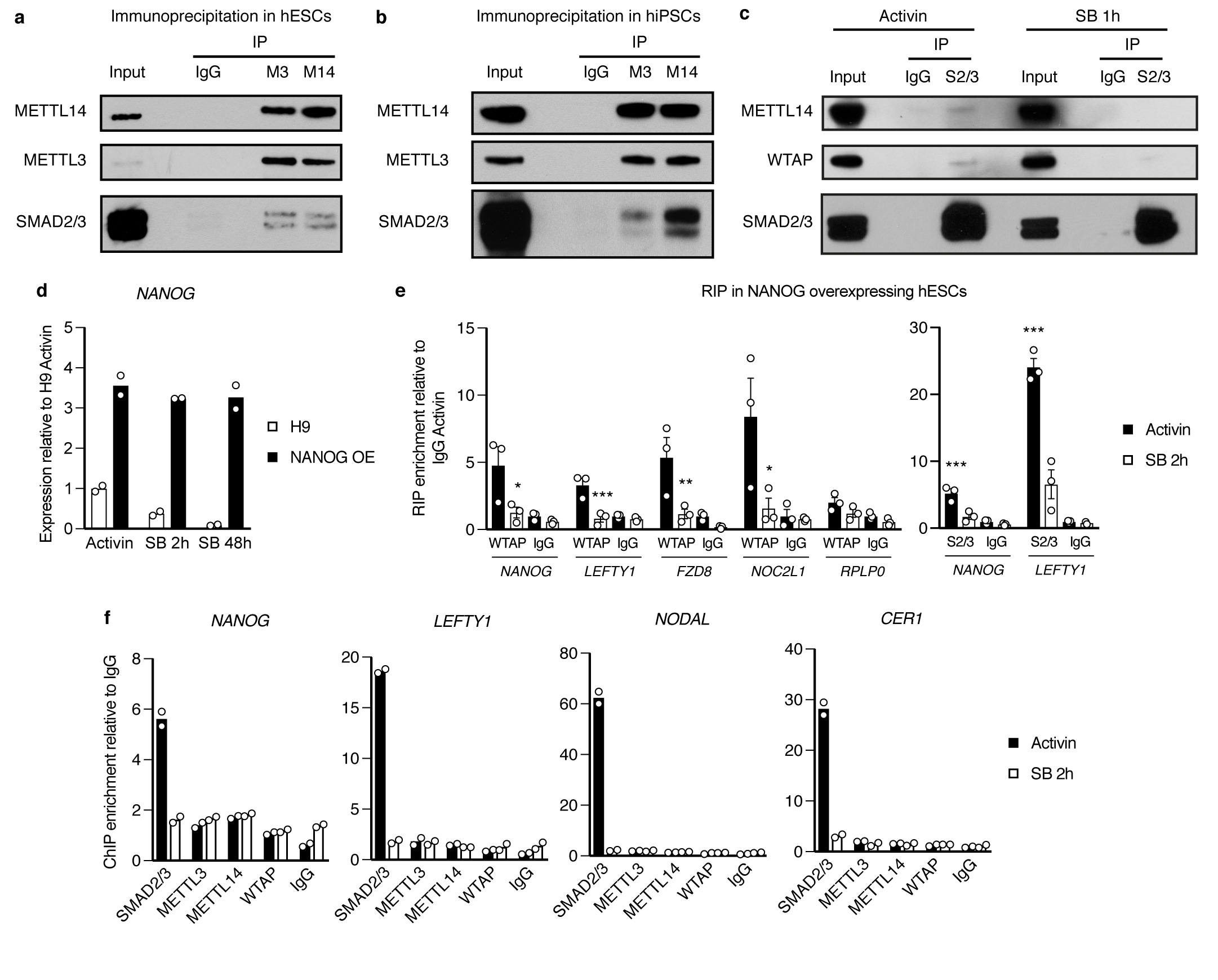
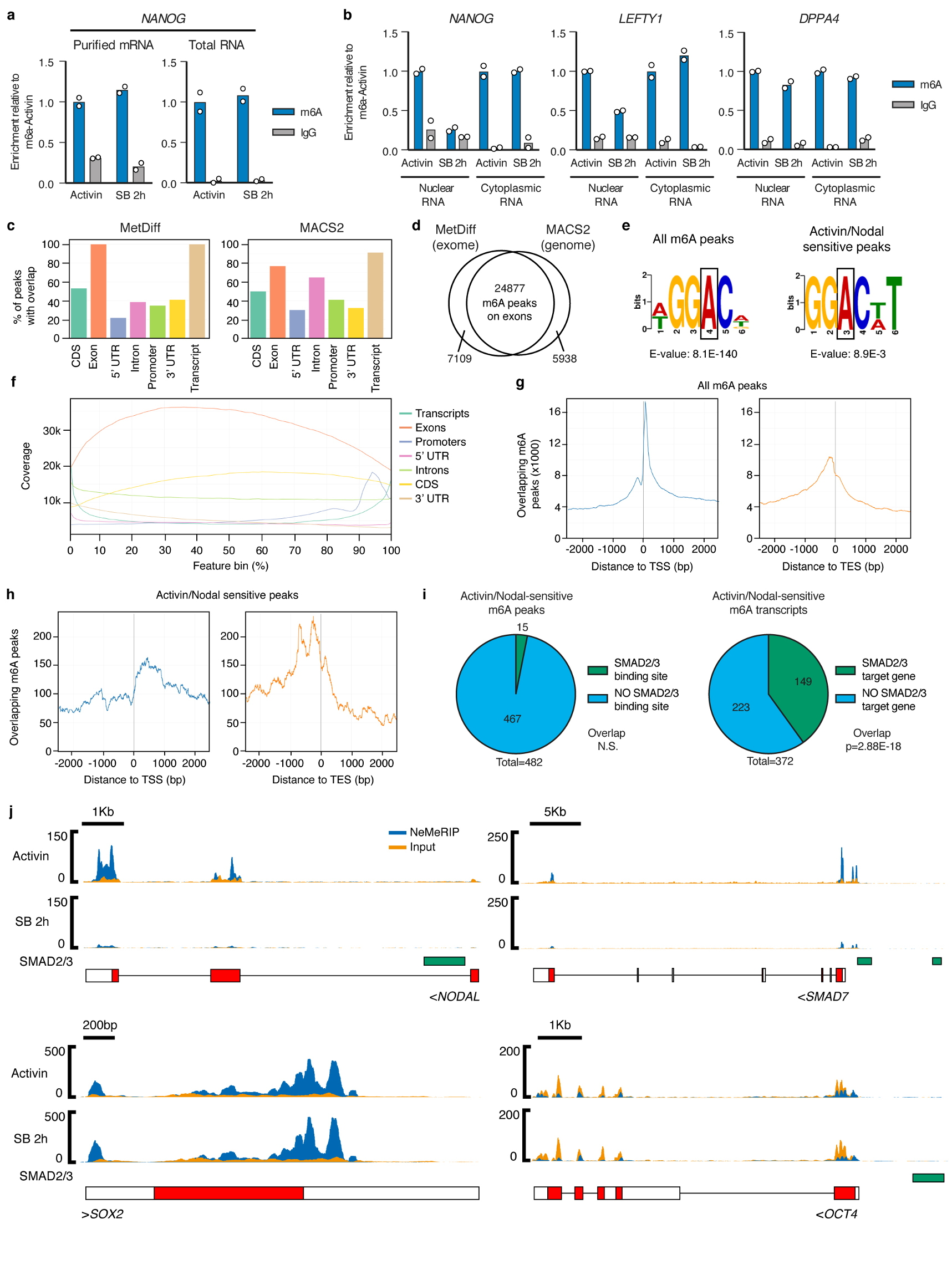
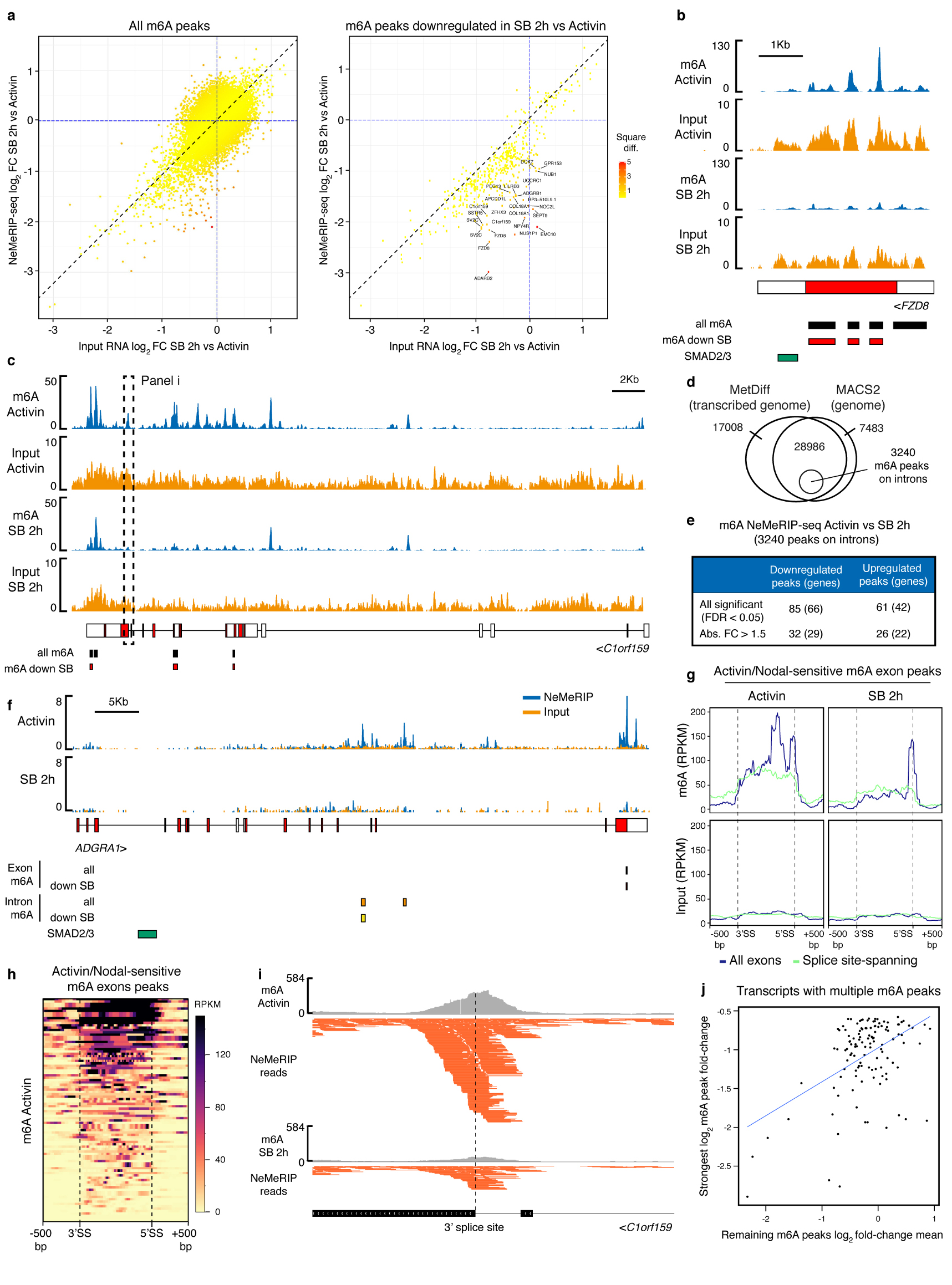
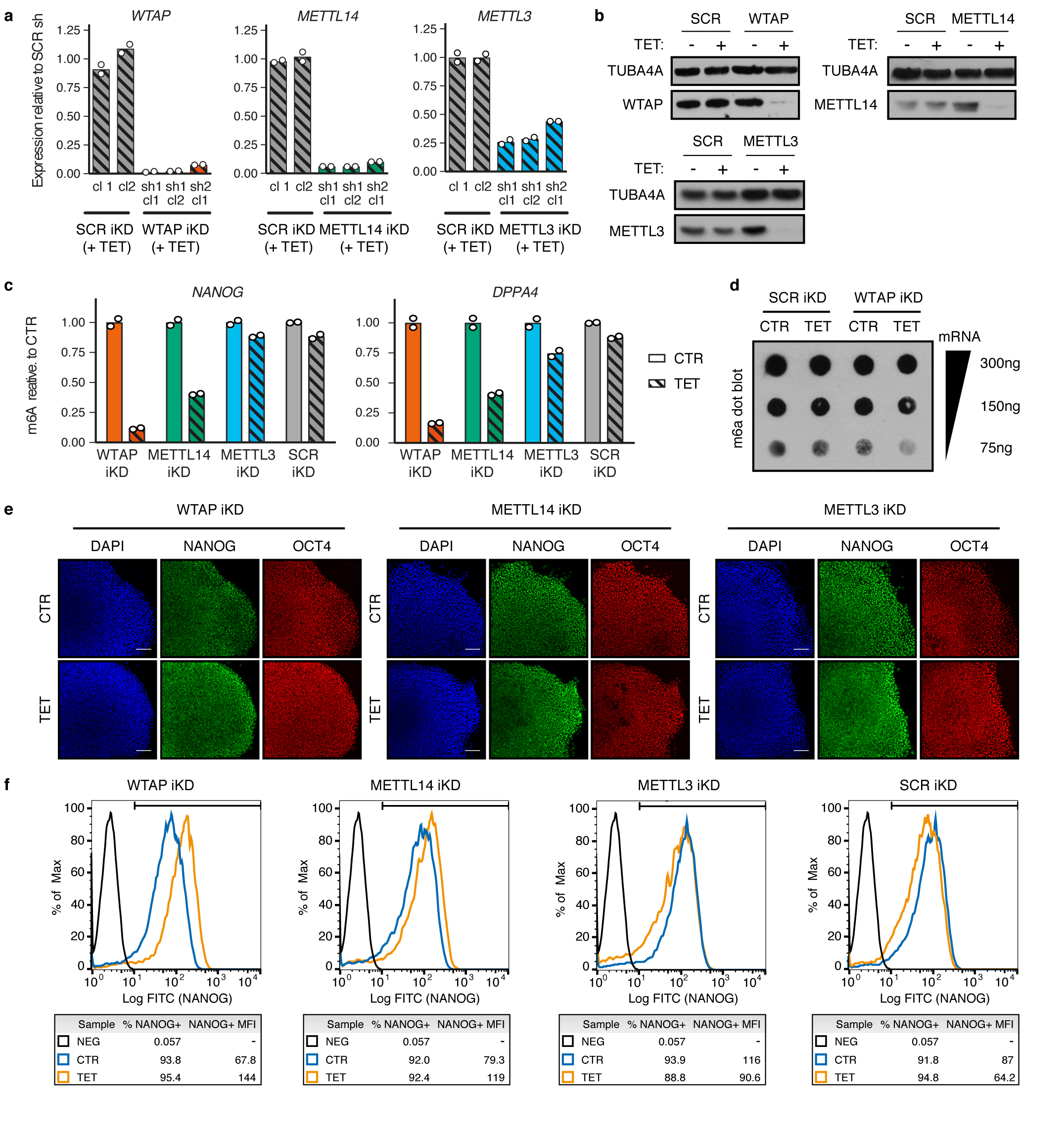
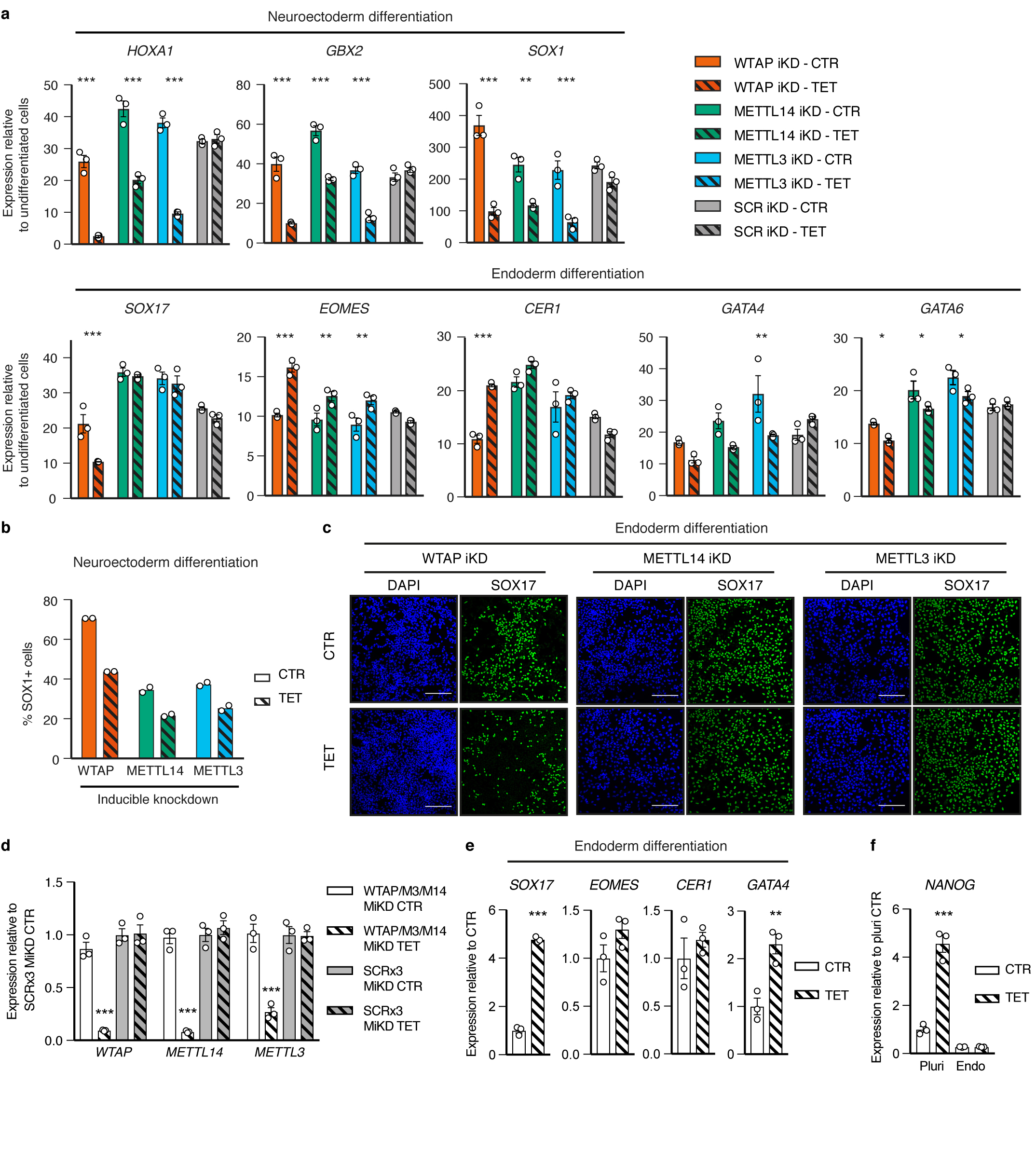
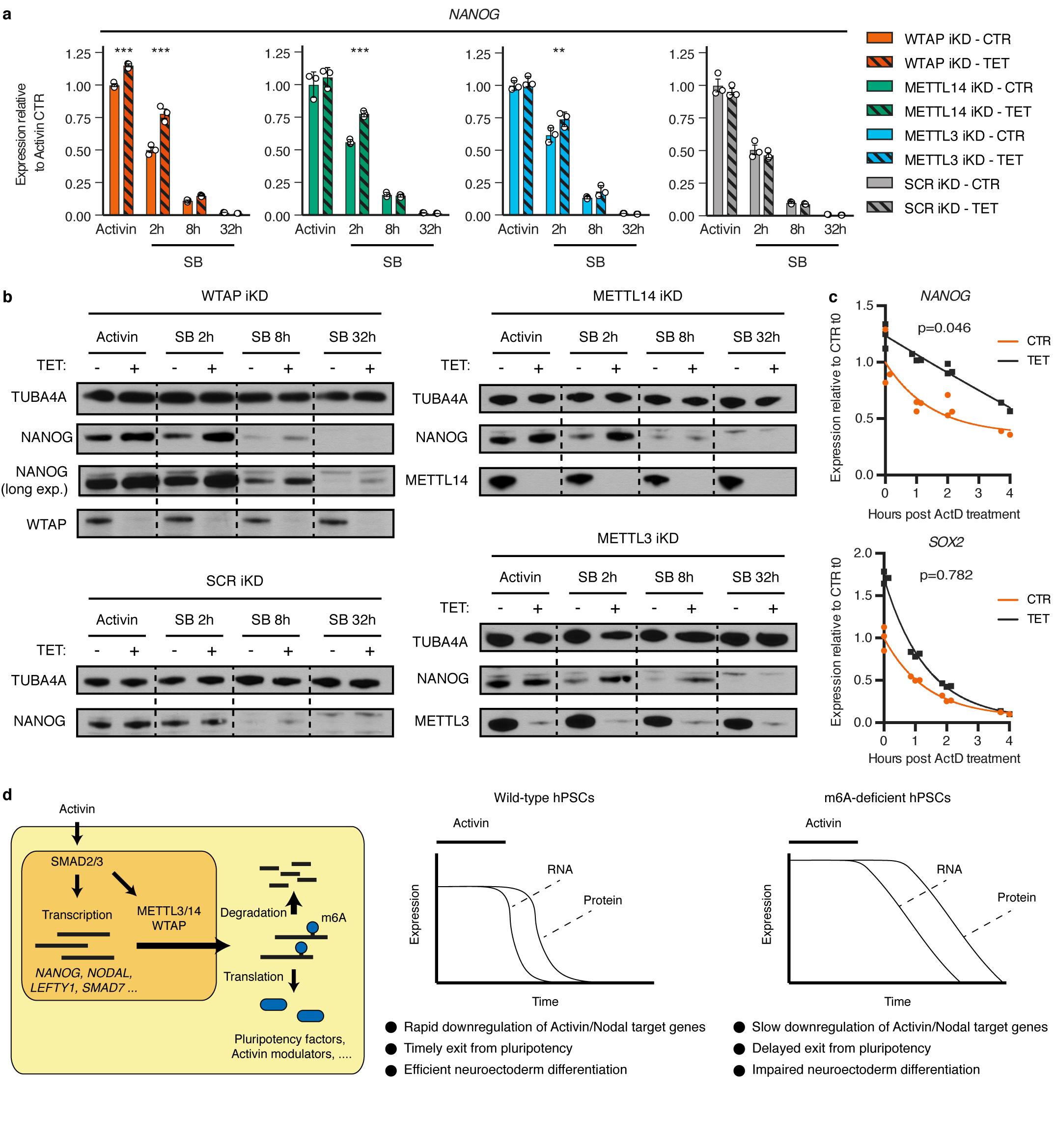
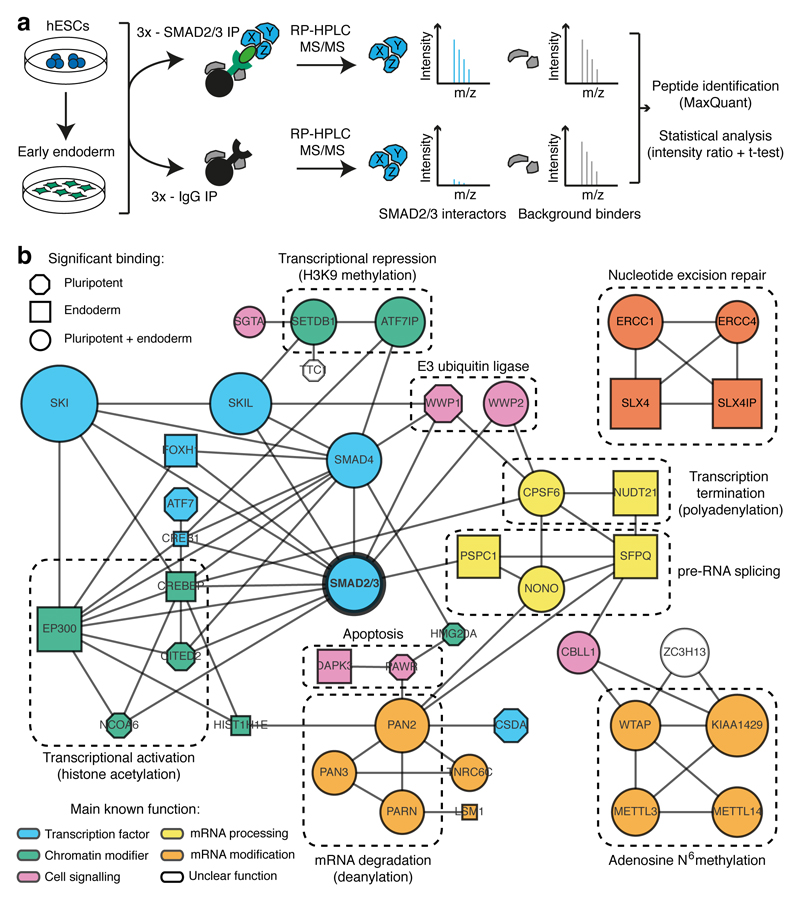
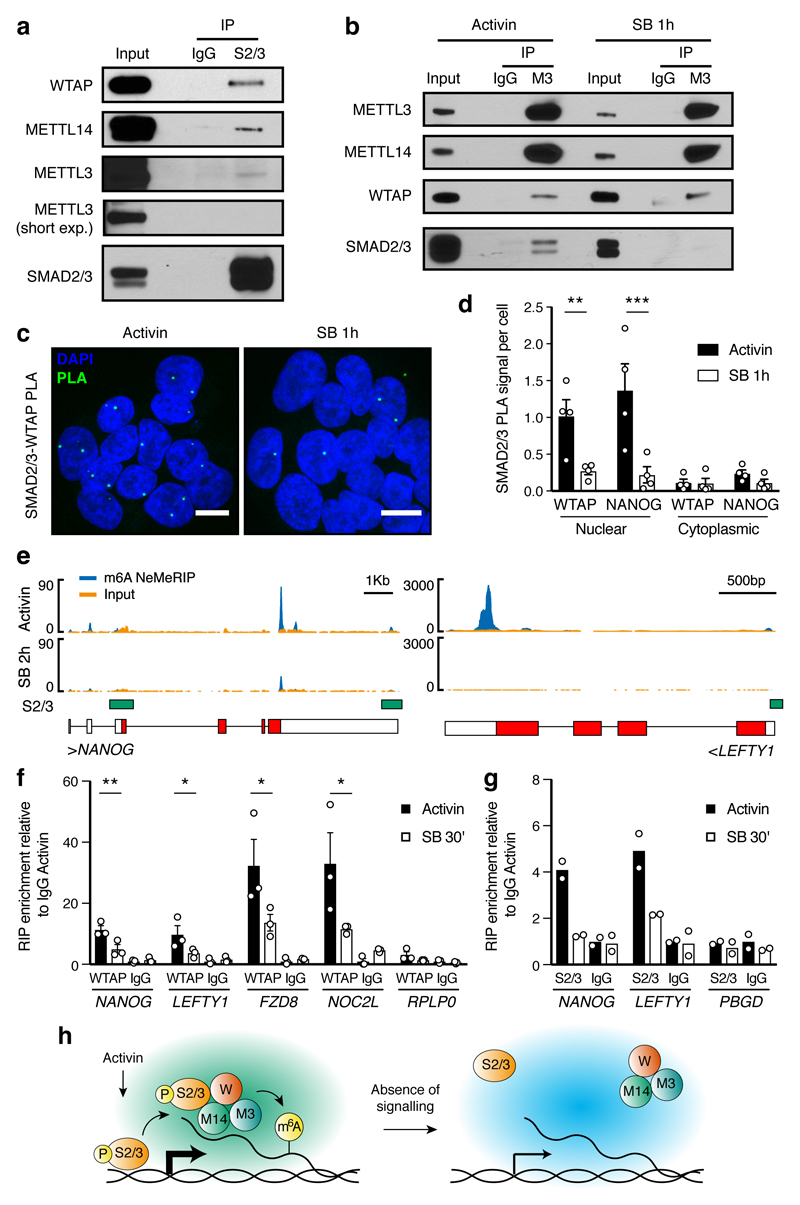
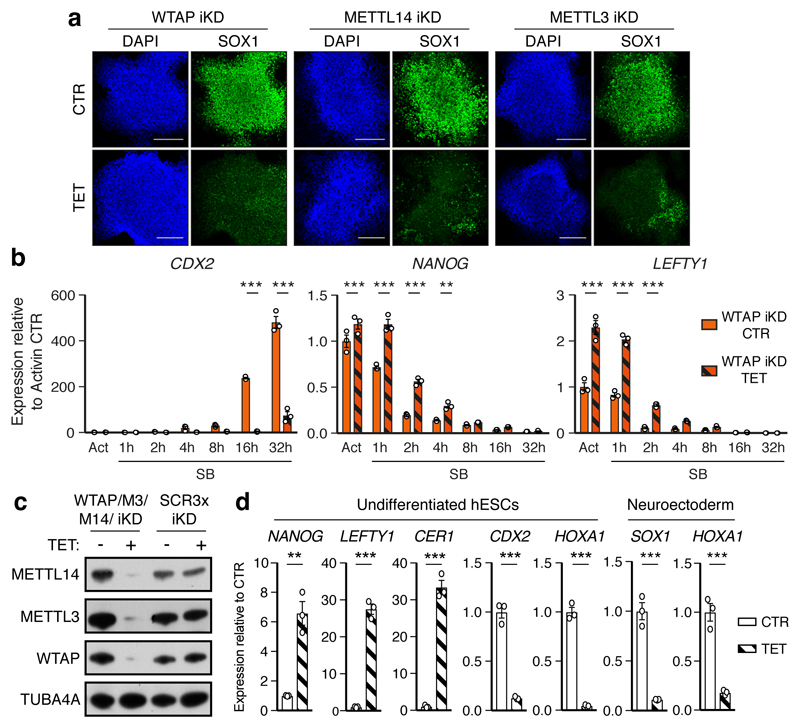
The TGFβ pathway has essential roles in embryonic development, organ homeostasis, tissue repair and disease. These diverse effects are mediated through the intracellular effectors SMAD2 and SMAD3 (hereafter SMAD2/3), whose canonical function is to control the activity of target genes by interacting with transcriptional regulators. Therefore, a complete description of the factors that interact with SMAD2/3 in a given cell type would have broad implications for many areas of cell biology. Here we describe the interactome of SMAD2/3 in human pluripotent stem cells. This analysis reveals that SMAD2/3 is involved in multiple molecular processes in addition to its role in transcription. In particular, we identify a functional interaction with the METTL3-METTL14-WTAP complex, which mediates the conversion of adenosine to N6-methyladenosine (m6A) on RNA. We show that SMAD2/3 promotes binding of the m6A methyltransferase complex to a subset of transcripts involved in early cell fate decisions. This mechanism destabilizes specific SMAD2/3 transcriptional targets, including the pluripotency factor gene NANOG, priming them for rapid downregulation upon differentiation to enable timely exit from pluripotency. Collectively, these findings reveal the mechanism by which extracellular signalling can induce rapid cellular responses through regulation of the epitranscriptome. These aspects of TGFβ signalling could have far-reaching implications in many other cell types and in diseases such as cancer.
Conflict of interest statement
No competing financial interests are declared.
Figures

Comment in
-
m6A deposition: a boost from TGFβ.Cell Res. 2018 May;28(5):505-506. doi: 10.1038/s41422-018-0037-3. Cell Res. 2018. PMID: 29679051 Free PMC article. No abstract available.
References
-
- Wu MY, Hill CS. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev Cell. 2009;16:329–43. - PubMed
-
- Gaarenstroom T, Hill CS. TGF-β signaling to chromatin: How Smads regulate transcription during self-renewal and differentiation. Semin Cell Dev Biol. 2014;32:107–8. - PubMed
-
- Heyn H, Esteller M. An Adenine Code for DNA: A Second Life for N6-Methyladenine. Cell. 2015;7:710–3. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
