

Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours
- PMID: 29489755
- PMCID: PMC5854542
- DOI: 10.1038/nature25795
Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours
Abstract
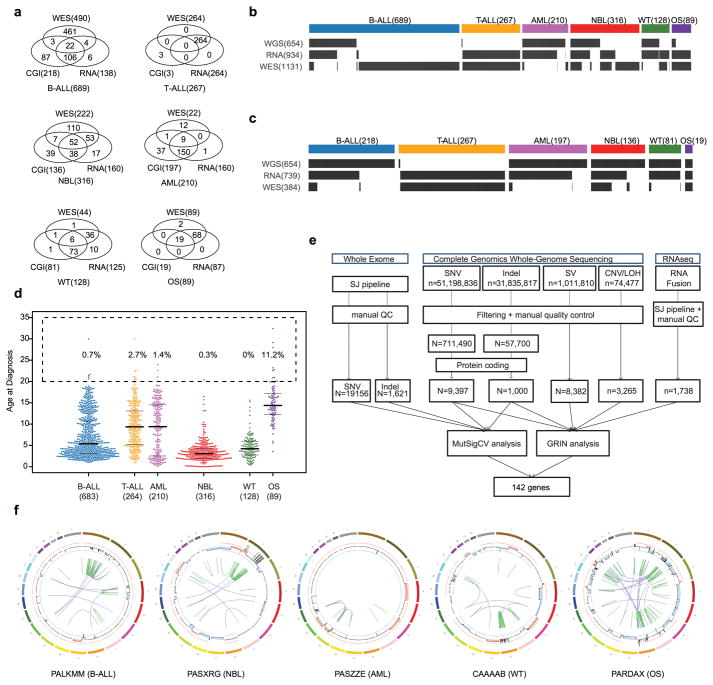
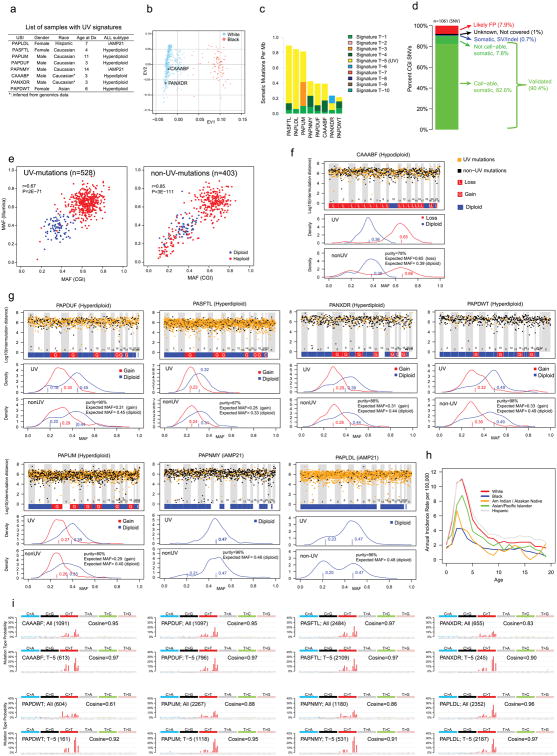
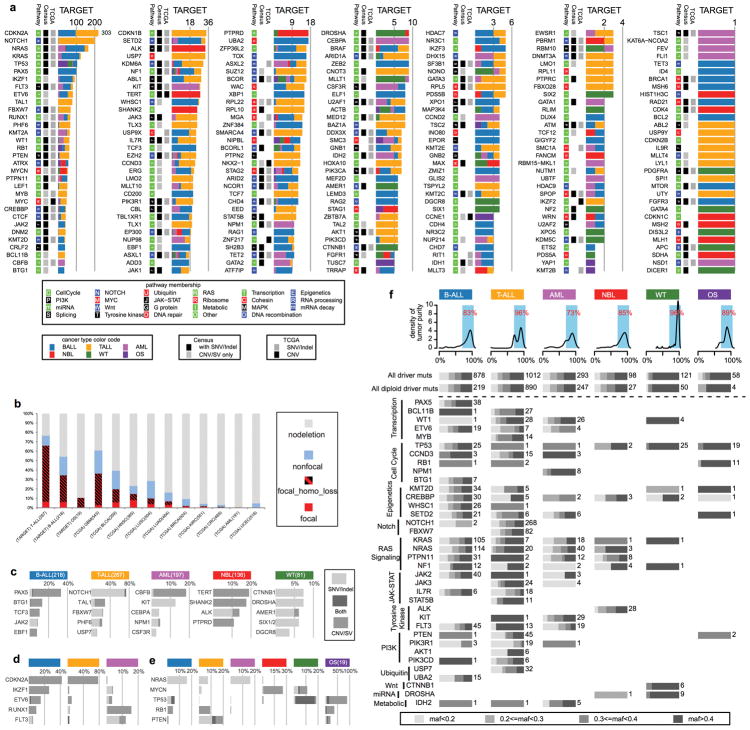
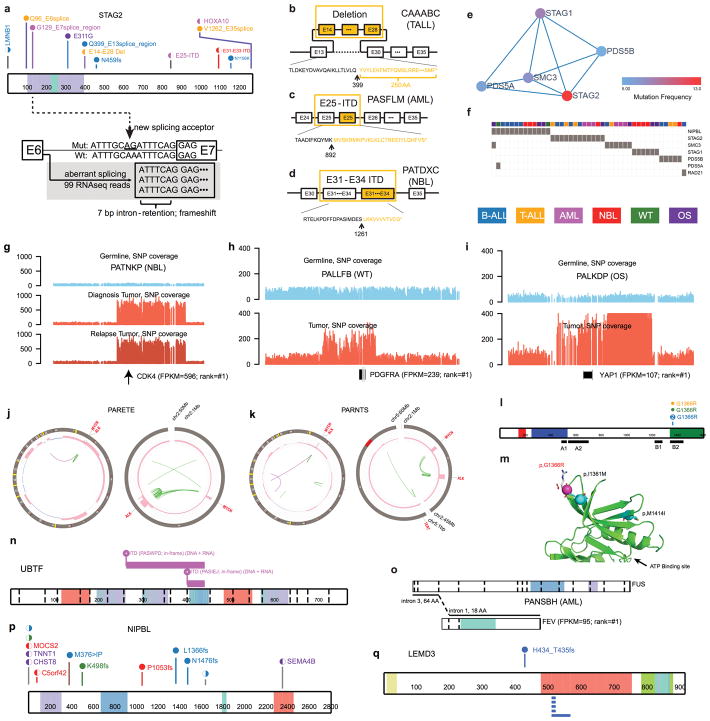
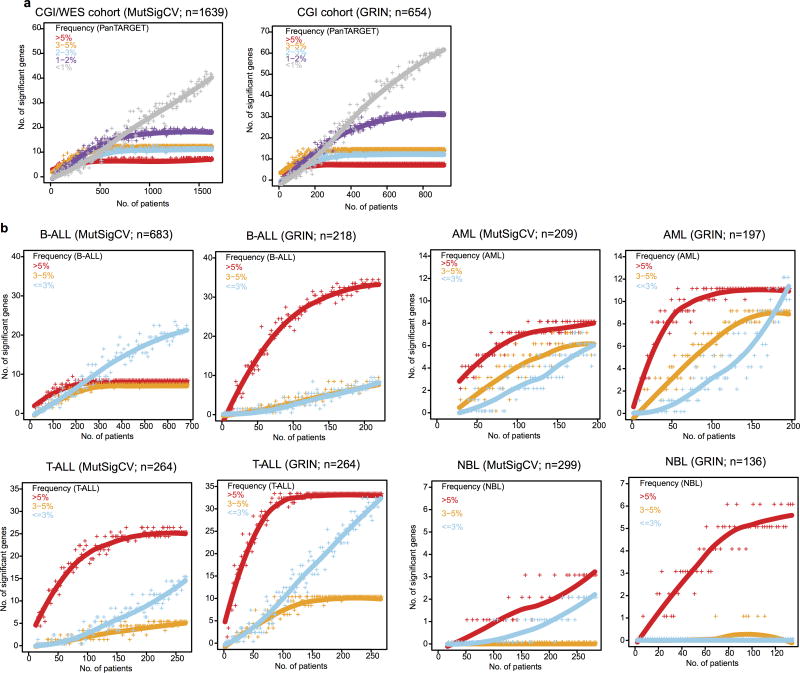
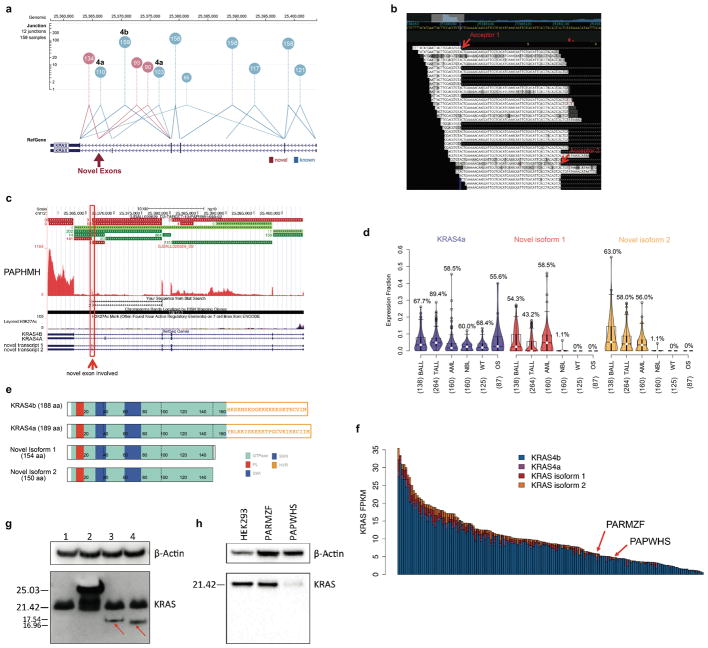
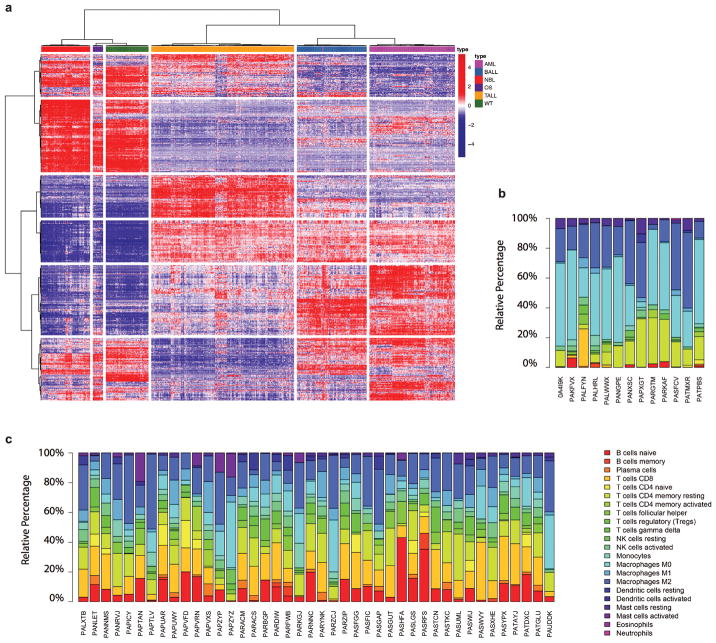
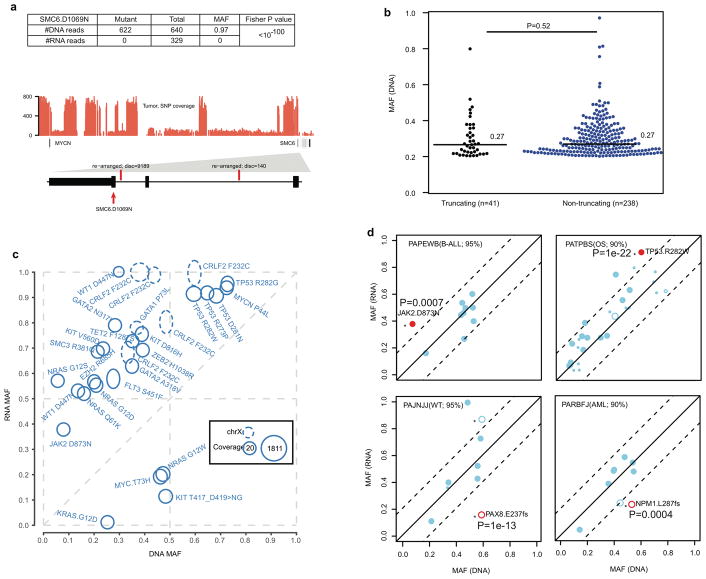
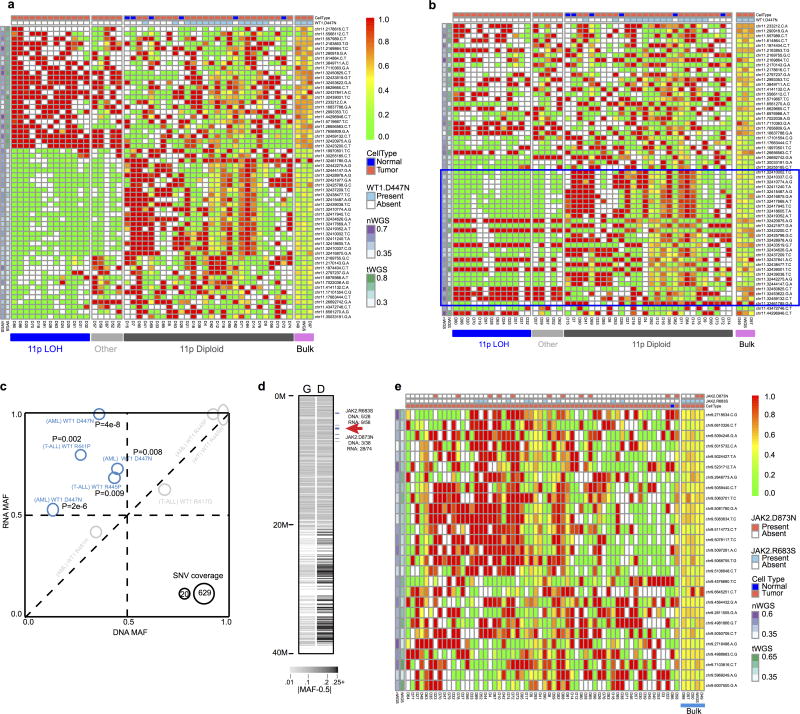
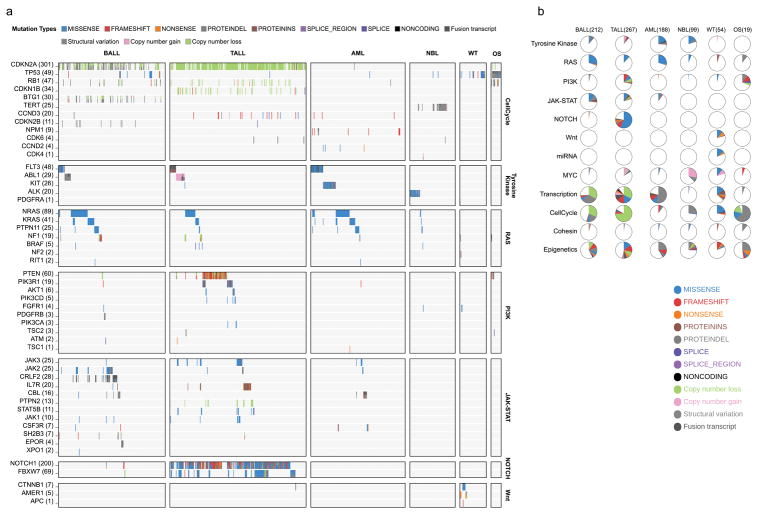
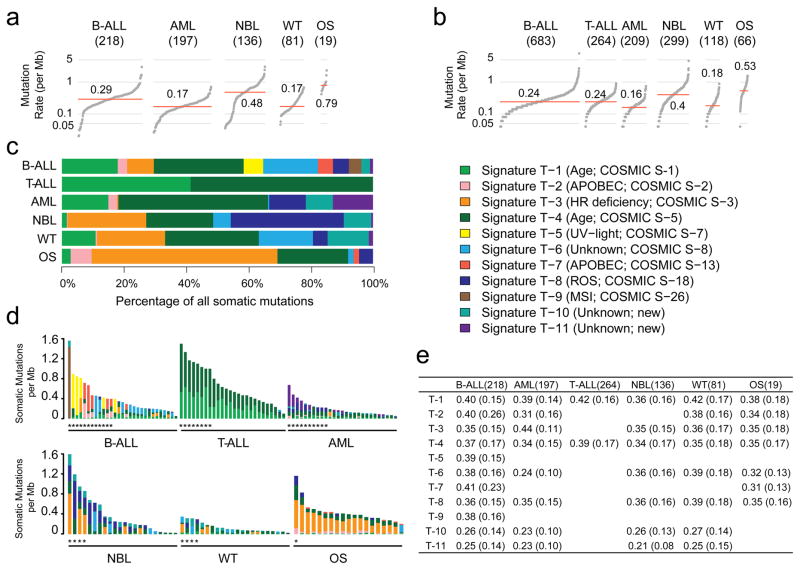
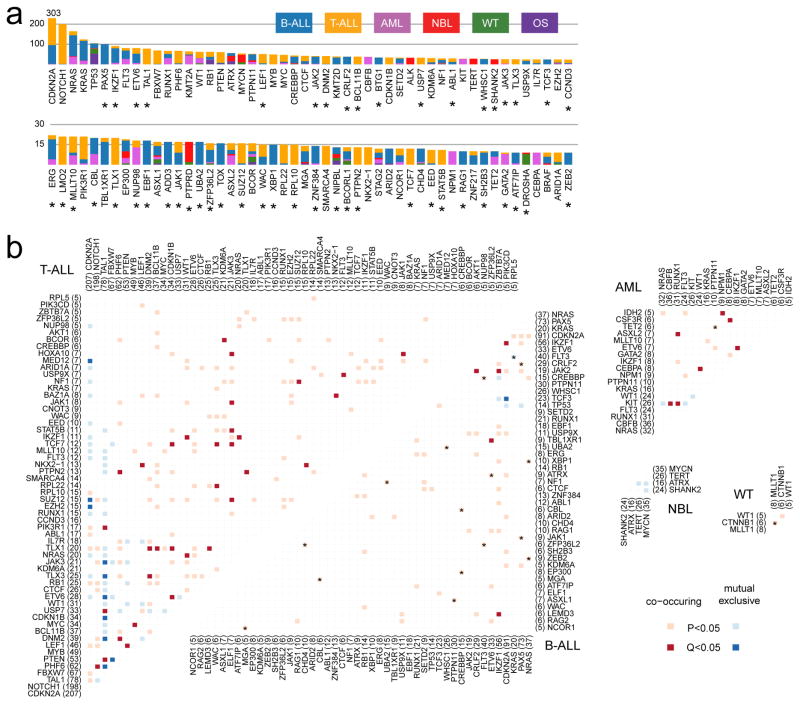
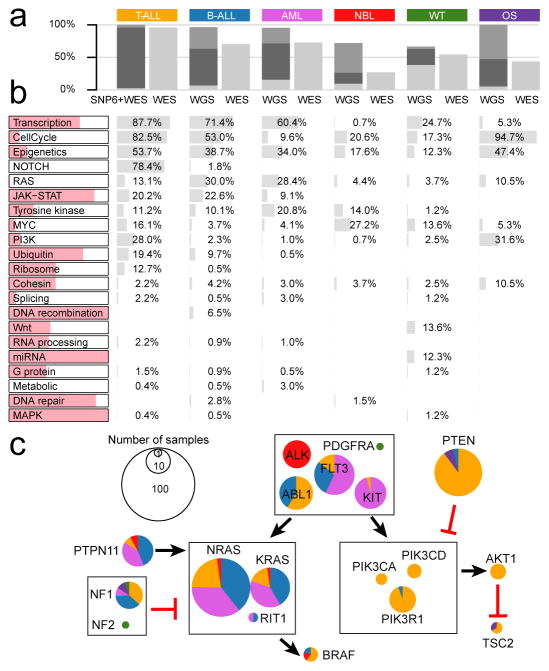
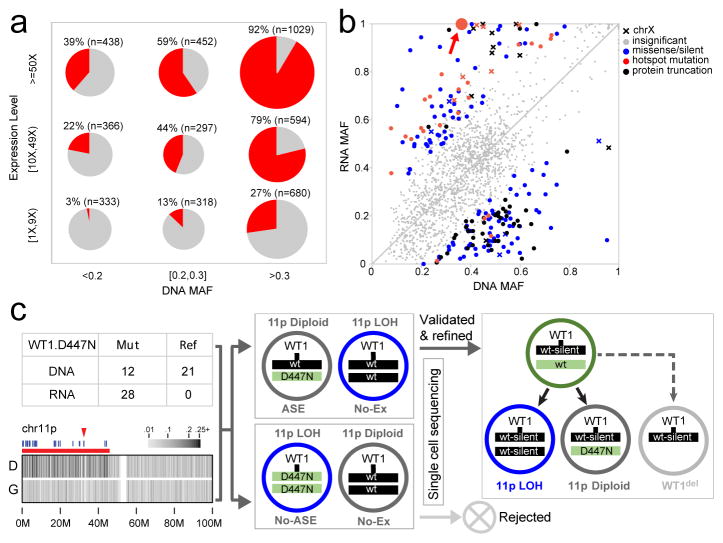
Analysis of molecular aberrations across multiple cancer types, known as pan-cancer analysis, identifies commonalities and differences in key biological processes that are dysregulated in cancer cells from diverse lineages. Pan-cancer analyses have been performed for adult but not paediatric cancers, which commonly occur in developing mesodermic rather than adult epithelial tissues. Here we present a pan-cancer study of somatic alterations, including single nucleotide variants, small insertions or deletions, structural variations, copy number alterations, gene fusions and internal tandem duplications in 1,699 paediatric leukaemias and solid tumours across six histotypes, with whole-genome, whole-exome and transcriptome sequencing data processed under a uniform analytical framework. We report 142 driver genes in paediatric cancers, of which only 45% match those found in adult pan-cancer studies; copy number alterations and structural variants constituted the majority (62%) of events. Eleven genome-wide mutational signatures were identified, including one attributed to ultraviolet-light exposure in eight aneuploid leukaemias. Transcription of the mutant allele was detectable for 34% of protein-coding mutations, and 20% exhibited allele-specific expression. These data provide a comprehensive genomic architecture for paediatric cancers and emphasize the need for paediatric cancer-specific development of precision therapies.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
New Insights into Genomics of Pediatric Cancers.Cancer Discov. 2018 May;8(5):OF6. doi: 10.1158/2159-8290.CD-NB2018-028. Epub 2018 Mar 12. Cancer Discov. 2018. PMID: 29530866
-
Landscapes of childhood tumours.Nature. 2018 Mar 15;555(7696):316-317. doi: 10.1038/d41586-018-01648-4. Nature. 2018. PMID: 29542703 No abstract available.
-
Paediatric cancer: Pan-cancer genomic analyses uncover molecular drivers.Nat Rev Clin Oncol. 2018 May;15(5):263. doi: 10.1038/nrclinonc.2018.43. Epub 2018 Mar 20. Nat Rev Clin Oncol. 2018. PMID: 29557392 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
