

Identification of Piperazinylbenzenesulfonamides as New Inhibitors of Claudin-1 Trafficking and Hepatitis C Virus Entry
- PMID: 29491159
- PMCID: PMC5923087
- DOI: 10.1128/JVI.01982-17
Identification of Piperazinylbenzenesulfonamides as New Inhibitors of Claudin-1 Trafficking and Hepatitis C Virus Entry
Abstract
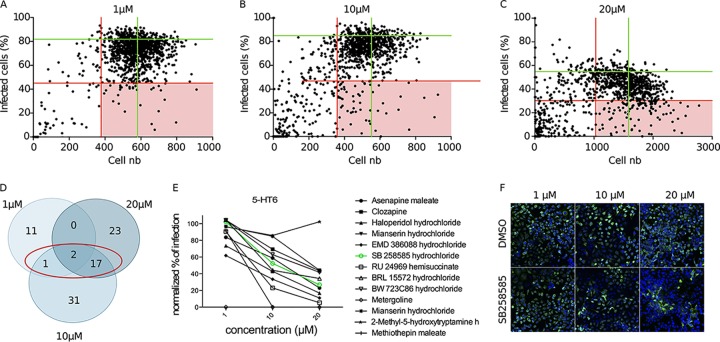
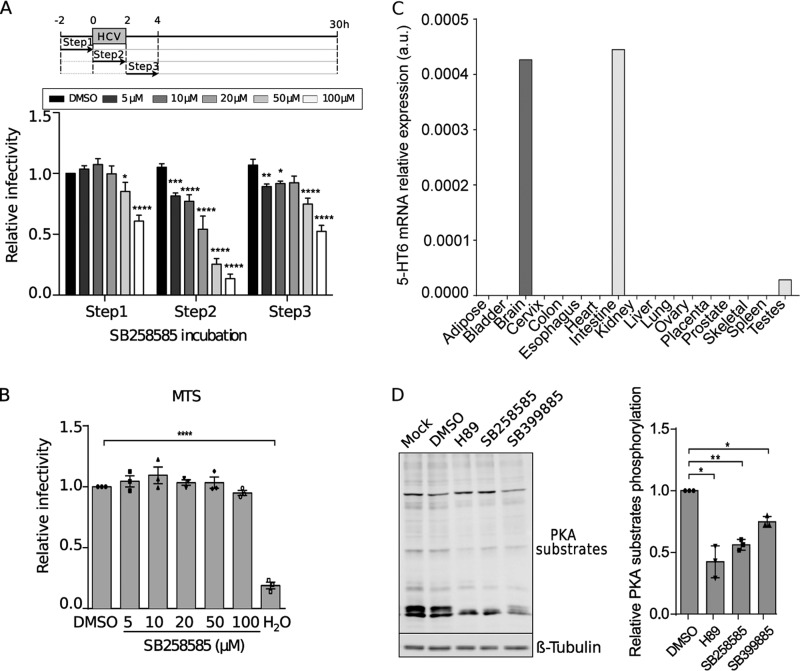
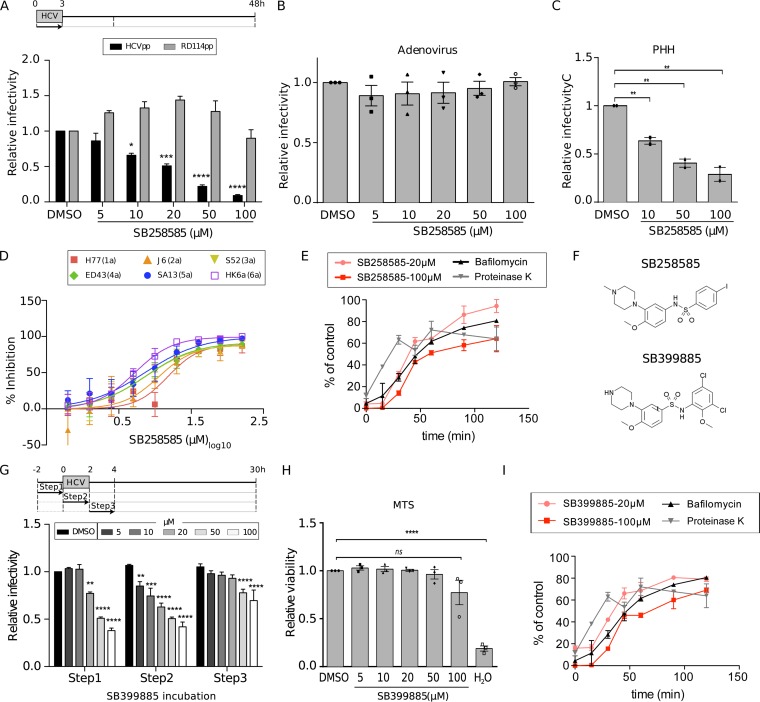
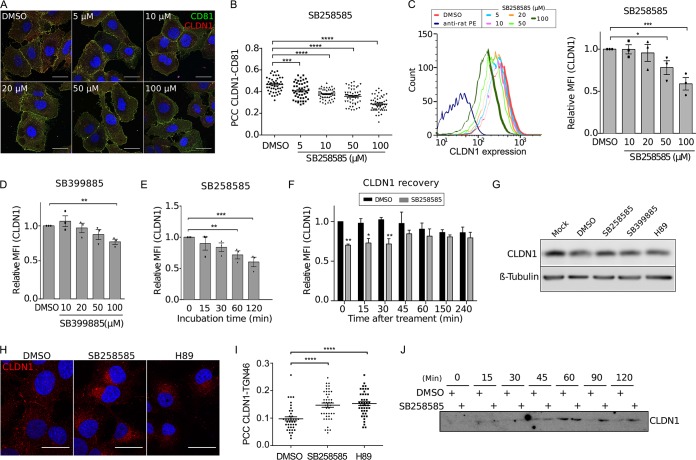
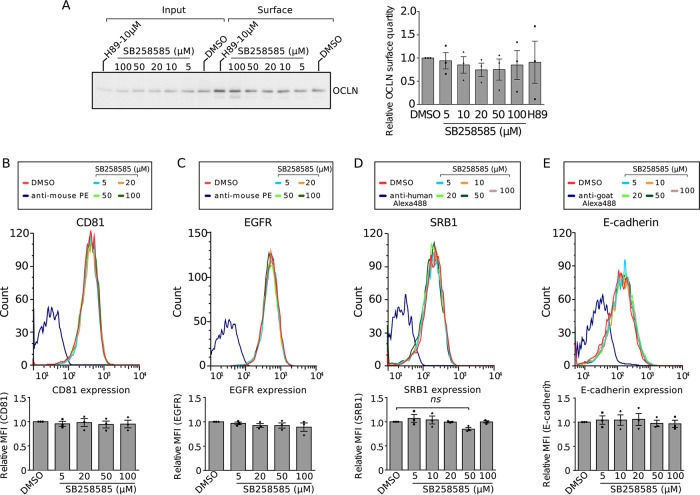
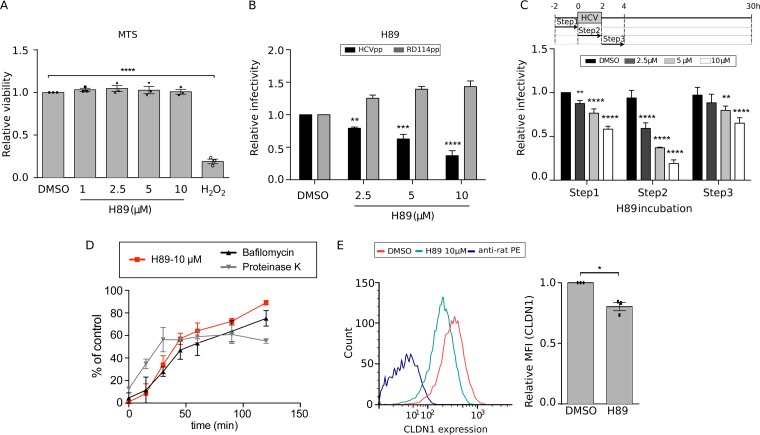
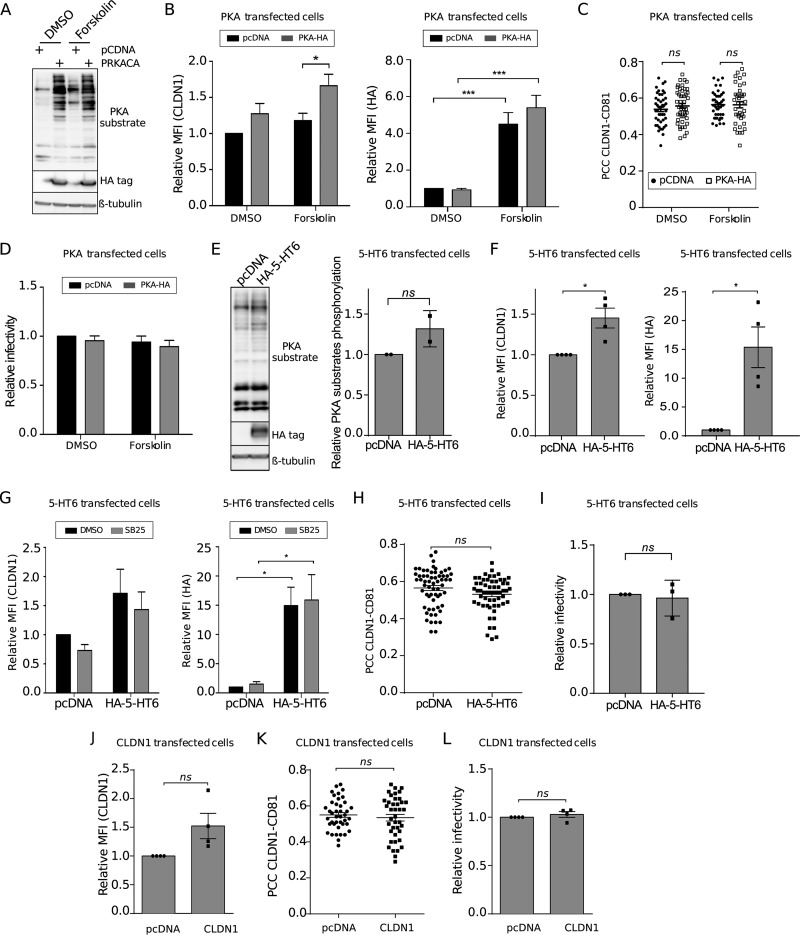
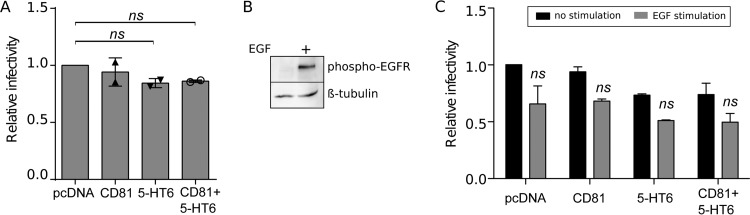
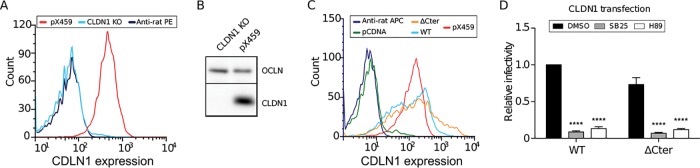
Hepatitis C virus (HCV) infection causes 500,000 deaths annually, in association with end-stage liver diseases. Investigations of the HCV life cycle have widened the knowledge of virology, and here we discovered that two piperazinylbenzenesulfonamides inhibit HCV entry into liver cells. The entry of HCV into host cells is a complex process that is not fully understood but is characterized by multiple spatially and temporally regulated steps involving several known host factors. Through a high-content virus infection screening analysis with a library of 1,120 biologically active chemical compounds, we identified SB258585, an antagonist of serotonin receptor 6 (5-HT6), as a new inhibitor of HCV entry in liver-derived cell lines as well as primary hepatocytes. A functional characterization suggested a role for this compound and the compound SB399885, which share similar structures, as inhibitors of a late HCV entry step, modulating the localization of the coreceptor tight junction protein claudin-1 (CLDN1) in a 5-HT6-independent manner. Both chemical compounds induced an intracellular accumulation of CLDN1, reflecting export impairment. This regulation correlated with the modulation of protein kinase A (PKA) activity. The PKA inhibitor H89 fully reproduced these phenotypes. Furthermore, PKA activation resulted in increased CLDN1 accumulation at the cell surface. Interestingly, an increase of CLDN1 recycling did not correlate with an increased interaction with CD81 or HCV entry. These findings reinforce the hypothesis of a common pathway, shared by several viruses, which involves G-protein-coupled receptor-dependent signaling in late steps of viral entry.IMPORTANCE The HCV entry process is highly complex, and important details of this structured event are poorly understood. By screening a library of biologically active chemical compounds, we identified two piperazinylbenzenesulfonamides as inhibitors of HCV entry. The mechanism of inhibition was not through the previously described activity of these inhibitors as antagonists of serotonin receptor 6 but instead through modulation of PKA activity in a 5-HT6-independent manner, as proven by the lack of 5-HT6 in the liver. We thus highlighted the involvement of the PKA pathway in modulating HCV entry at a postbinding step and in the recycling of the tight junction protein claudin-1 (CLDN1) toward the cell surface. Our work underscores once more the complexity of HCV entry steps and suggests a role for the PKA pathway as a regulator of CLDN1 recycling, with impacts on both cell biology and virology.
Keywords: hepatitis C virus; piperazinylbenzenesulfonamide; protein kinase A; tight junction protein; virus entry; virus-host interaction.
Copyright © 2018 American Society for Microbiology.
Figures
Similar articles
-
Protein kinase A-dependent step(s) in hepatitis C virus entry and infectivity.J Virol. 2008 Sep;82(17):8797-811. doi: 10.1128/JVI.00592-08. Epub 2008 Jun 25. J Virol. 2008. PMID: 18579596 Free PMC article.
-
Polarization restricts hepatitis C virus entry into HepG2 hepatoma cells.J Virol. 2009 Jun;83(12):6211-21. doi: 10.1128/JVI.00246-09. Epub 2009 Apr 8. J Virol. 2009. PMID: 19357163 Free PMC article.
-
Infection with hepatitis C virus depends on TACSTD2, a regulator of claudin-1 and occludin highly downregulated in hepatocellular carcinoma.PLoS Pathog. 2018 Mar 14;14(3):e1006916. doi: 10.1371/journal.ppat.1006916. eCollection 2018 Mar. PLoS Pathog. 2018. PMID: 29538454 Free PMC article.
-
How hepatitis C virus invades hepatocytes: the mystery of viral entry.World J Gastroenterol. 2014 Apr 7;20(13):3457-67. doi: 10.3748/wjg.v20.i13.3457. World J Gastroenterol. 2014. PMID: 24707128 Free PMC article. Review.
-
Hepatitis C Virus Entry: Protein Interactions and Fusion Determinants Governing Productive Hepatocyte Invasion.Cold Spring Harb Perspect Med. 2020 Feb 3;10(2):a036830. doi: 10.1101/cshperspect.a036830. Cold Spring Harb Perspect Med. 2020. PMID: 31427285 Free PMC article. Review.
Cited by
-
A network biology approach to identify crucial host targets for COVID-19.Methods. 2022 Jul;203:108-115. doi: 10.1016/j.ymeth.2022.03.016. Epub 2022 Mar 29. Methods. 2022. PMID: 35364279 Free PMC article.
-
Variations in maternal adenylate cyclase genes are associated with congenital Zika syndrome in a cohort from Northeast, Brazil.J Intern Med. 2019 Feb;285(2):215-222. doi: 10.1111/joim.12829. Epub 2018 Sep 17. J Intern Med. 2019. PMID: 30222212 Free PMC article.
-
Hepatitis C Virus Entry: An Intriguingly Complex and Highly Regulated Process.Int J Mol Sci. 2020 Mar 18;21(6):2091. doi: 10.3390/ijms21062091. Int J Mol Sci. 2020. PMID: 32197477 Free PMC article. Review.
-
Hepatitis C virus infection and tight junction proteins: The ties that bind.Biochim Biophys Acta Biomembr. 2020 Jul 1;1862(7):183296. doi: 10.1016/j.bbamem.2020.183296. Epub 2020 Apr 5. Biochim Biophys Acta Biomembr. 2020. PMID: 32268133 Free PMC article. Review.
-
Hypervariable Region 1 in Envelope Protein 2 of Hepatitis C Virus: A Linchpin in Neutralizing Antibody Evasion and Viral Entry.Front Immunol. 2018 Sep 27;9:2146. doi: 10.3389/fimmu.2018.02146. eCollection 2018. Front Immunol. 2018. PMID: 30319614 Free PMC article. Review.
References
-
- Potel J, Rassam P, Montpellier C, Kaestner L, Werkmeister E, Tews BA, Couturier C, Popescu C-I, Baumert TF, Rubinstein E, Dubuisson J, Milhiet P-E, Cocquerel L. 2013. EWI-2wint promotes CD81 clustering that abrogates hepatitis C virus entry: dynamics and partitioning of CD81. Cell Microbiol 15:1234–1252. doi: 10.1111/cmi.12112. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
