

Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells
- PMID: 29491378
- PMCID: PMC5830451
- DOI: 10.1038/s41467-018-03367-w
Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells
Abstract
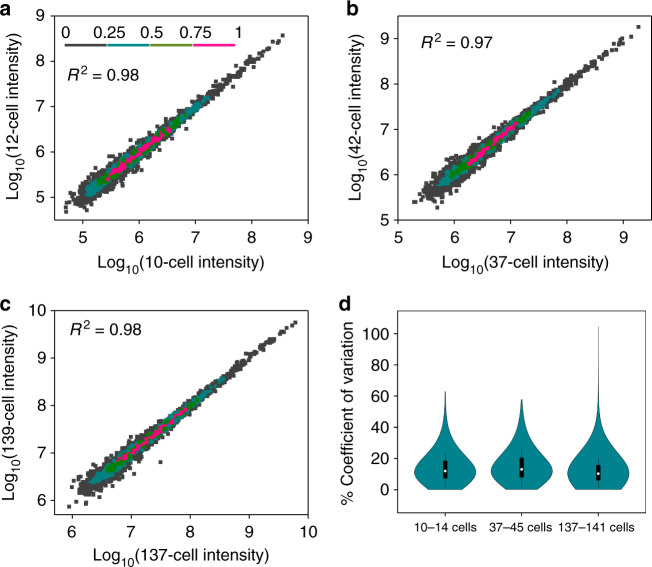
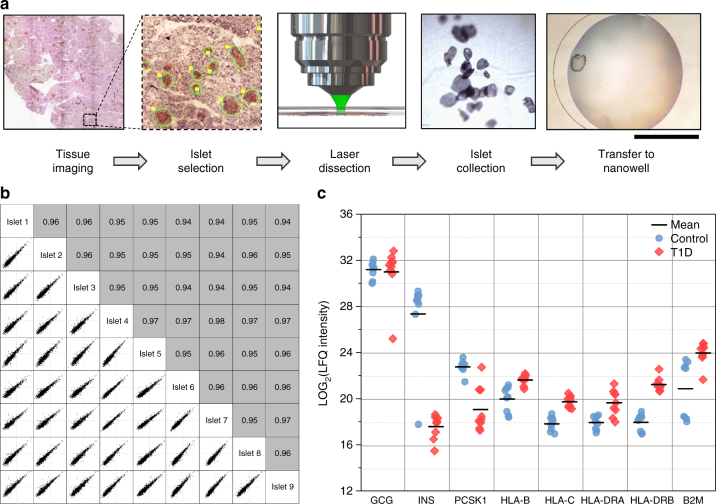
Nanoscale or single-cell technologies are critical for biomedical applications. However, current mass spectrometry (MS)-based proteomic approaches require samples comprising a minimum of thousands of cells to provide in-depth profiling. Here, we report the development of a nanoPOTS (nanodroplet processing in one pot for trace samples) platform for small cell population proteomics analysis. NanoPOTS enhances the efficiency and recovery of sample processing by downscaling processing volumes to <200 nL to minimize surface losses. When combined with ultrasensitive liquid chromatography-MS, nanoPOTS allows identification of ~1500 to ~3000 proteins from ~10 to ~140 cells, respectively. By incorporating the Match Between Runs algorithm of MaxQuant, >3000 proteins are consistently identified from as few as 10 cells. Furthermore, we demonstrate quantification of ~2400 proteins from single human pancreatic islet thin sections from type 1 diabetic and control donors, illustrating the application of nanoPOTS for spatially resolved proteome measurements from clinical tissues.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
