

A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease
- PMID: 29491396
- PMCID: PMC5832428
- DOI: 10.1038/s41419-017-0215-0
A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease
Abstract
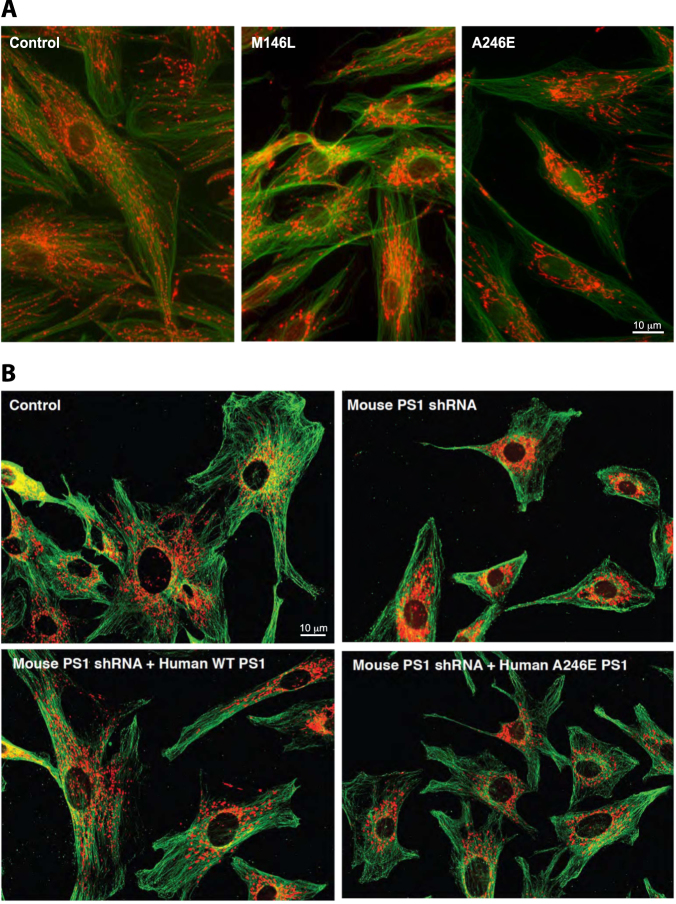
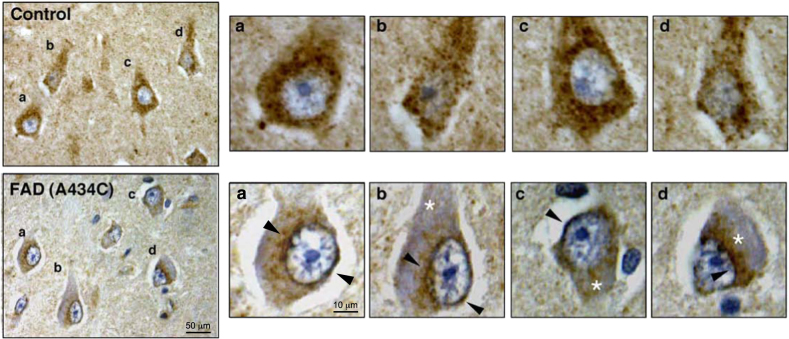
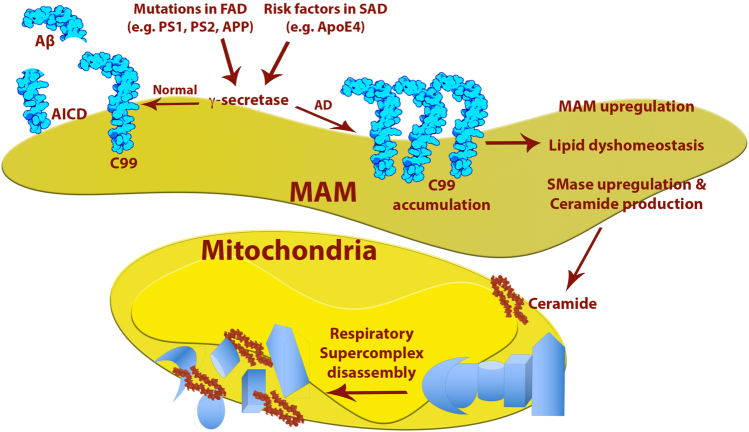
In the last few years, increased emphasis has been devoted to understanding the contribution of mitochondria-associated endoplasmic reticulum (ER) membranes (MAM) to human pathology in general, and neurodegenerative diseases in particular. A major reason for this is the central role that this subdomain of the ER plays in metabolic regulation and in mitochondrial biology. As such, aberrant MAM function may help explain the seemingly unrelated metabolic abnormalities often seen in neurodegeneration. In the specific case of Alzheimer disease (AD), besides perturbations in calcium and lipid homeostasis, there are numerous documented alterations in mitochondrial behavior and function, including reduced respiratory chain activity and oxidative phosphorylation, increased free radical production, and altered organellar morphology, dynamics, and positioning (especially perinuclear mitochondria). However, whether these alterations are primary events causative of the disease, or are secondary downstream events that are the result of some other, more fundamental problem, is still unclear. In support of the former possibility, we recently reported that C99, the C-terminal processing product of the amyloid precursor protein (APP) derived from its cleavage by β-secretase, is present in MAM, that its level is increased in AD, and that this increase reduces mitochondrial respiration, likely via a C99-induced alteration in cellular sphingolipid homeostasis. Thus, the metabolic disturbances seen in AD likely arise from increased ER-mitochondrial communication that is driven by an increase in the levels of C99 at the MAM.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
