

Absence of Claudin 11 in CNS Myelin Perturbs Behavior and Neurotransmitter Levels in Mice
- PMID: 29491447
- PMCID: PMC5830493
- DOI: 10.1038/s41598-018-22047-9
Absence of Claudin 11 in CNS Myelin Perturbs Behavior and Neurotransmitter Levels in Mice
Abstract
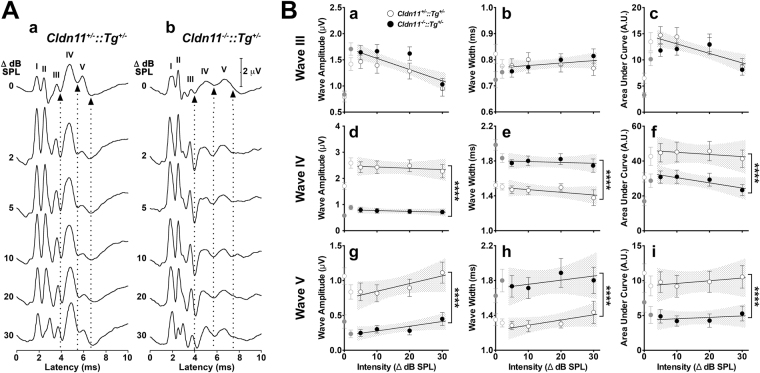
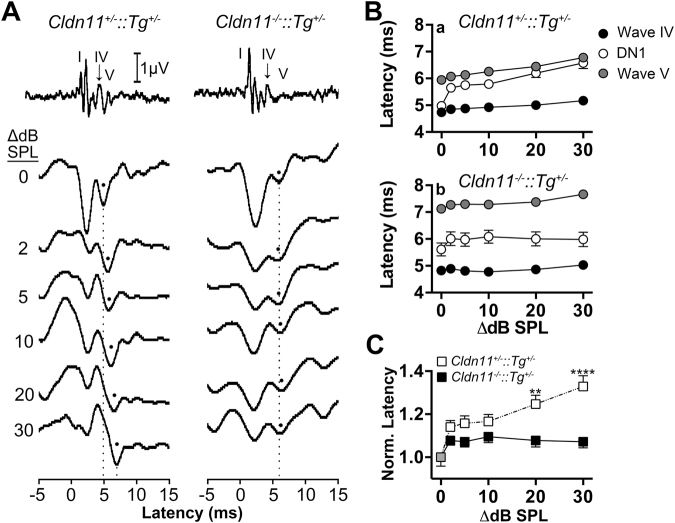
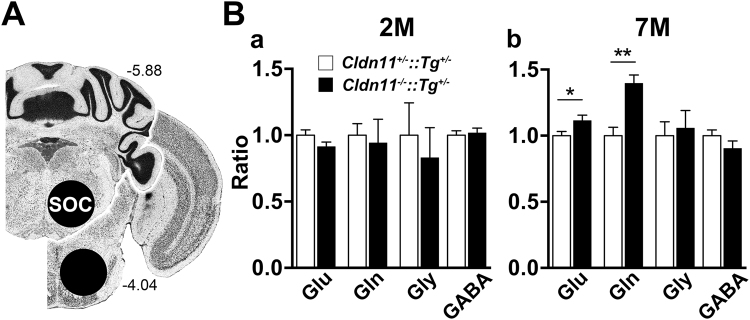
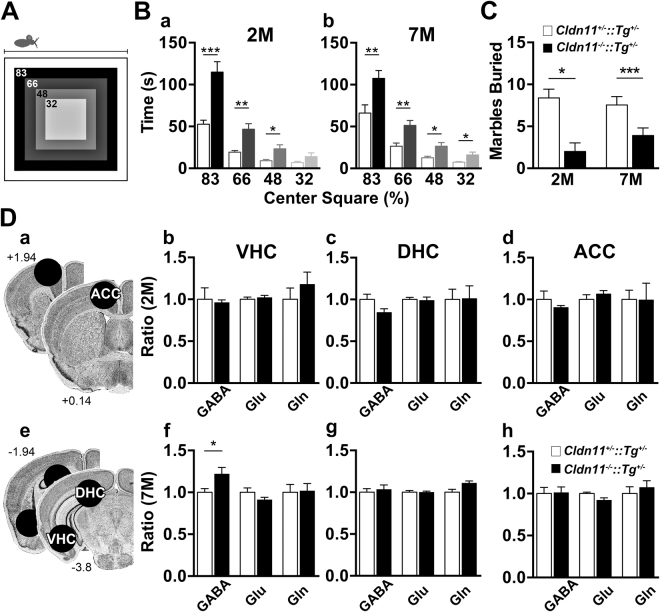
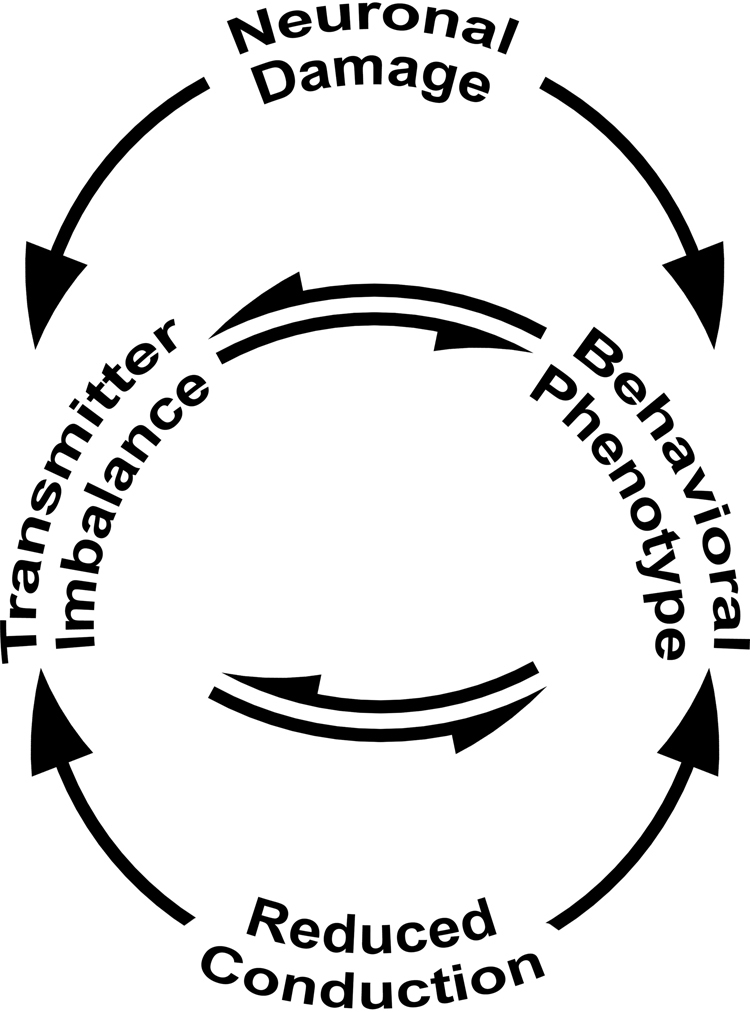
Neuronal origins of behavioral disorders have been examined for decades to construct frameworks for understanding psychiatric diseases and developing useful therapeutic strategies with clinical application. Despite abundant anecdotal evidence for white matter etiologies, including altered tractography in neuroimaging and diminished oligodendrocyte-specific gene expression in autopsy studies, mechanistic data demonstrating that dysfunctional myelin sheaths can cause behavioral deficits and perturb neurotransmitter biochemistry have not been forthcoming. At least in part, this impasse stems from difficulties in identifying model systems free of degenerative pathology to enable unambiguous assessment of neuron biology and behavior in a background of myelin dysfunction. Herein we examine myelin mutant mice lacking expression of the Claudin11 gene in oligodendrocytes and characterize two behavioral endophenotypes: perturbed auditory processing and reduced anxiety/avoidance. Importantly, these behaviors are associated with increased transmission time along myelinated fibers as well as glutamate and GABA neurotransmitter imbalances in auditory brainstem and amygdala, in the absence of neurodegeneration. Thus, our findings broaden the etiology of neuropsychiatric disease to include dysfunctional myelin, and identify a preclinical model for the development of novel disease-modifying therapies.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Flagstad P, et al. Disruption of neurogenesis on gestational day 17 in the rat causes behavioral changes relevant to positive and negative schizophrenia symptoms and alters amphetamine-induced dopamine release in nucleus accumbens. Neuropsychopharmacology. 2004;29:2052–2064. doi: 10.1038/sj.npp.1300516. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
