

Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online
- PMID: 29494574
- PMCID: PMC5937130
- DOI: 10.1038/nprot.2017.151
Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online
Abstract
Systems biology is the study of complex living organisms, and as such, analysis on a systems-wide scale involves the collection of information-dense data sets that are representative of an entire phenotype. To uncover dynamic biological mechanisms, bioinformatics tools have become essential to facilitating data interpretation in large-scale analyses. Global metabolomics is one such method for performing systems biology, as metabolites represent the downstream functional products of ongoing biological processes. We have developed XCMS Online, a platform that enables online metabolomics data processing and interpretation. A systems biology workflow recently implemented within XCMS Online enables rapid metabolic pathway mapping using raw metabolomics data for investigating dysregulated metabolic processes. In addition, this platform supports integration of multi-omic (such as genomic and proteomic) data to garner further systems-wide mechanistic insight. Here, we provide an in-depth procedure showing how to effectively navigate and use the systems biology workflow within XCMS Online without a priori knowledge of the platform, including uploading liquid chromatography (LC)-mass spectrometry (MS) data from metabolite-extracted biological samples, defining the job parameters to identify features, correcting for retention time deviations, conducting statistical analysis of features between sample classes and performing predictive metabolic pathway analysis. Additional multi-omics data can be uploaded and overlaid with previously identified pathways to enhance systems-wide analysis of the observed dysregulations. We also describe unique visualization tools to assist in elucidation of statistically significant dysregulated metabolic pathways. Parameter input takes 5-10 min, depending on user experience; data processing typically takes 1-3 h, and data analysis takes ∼30 min.
Conflict of interest statement
Figures
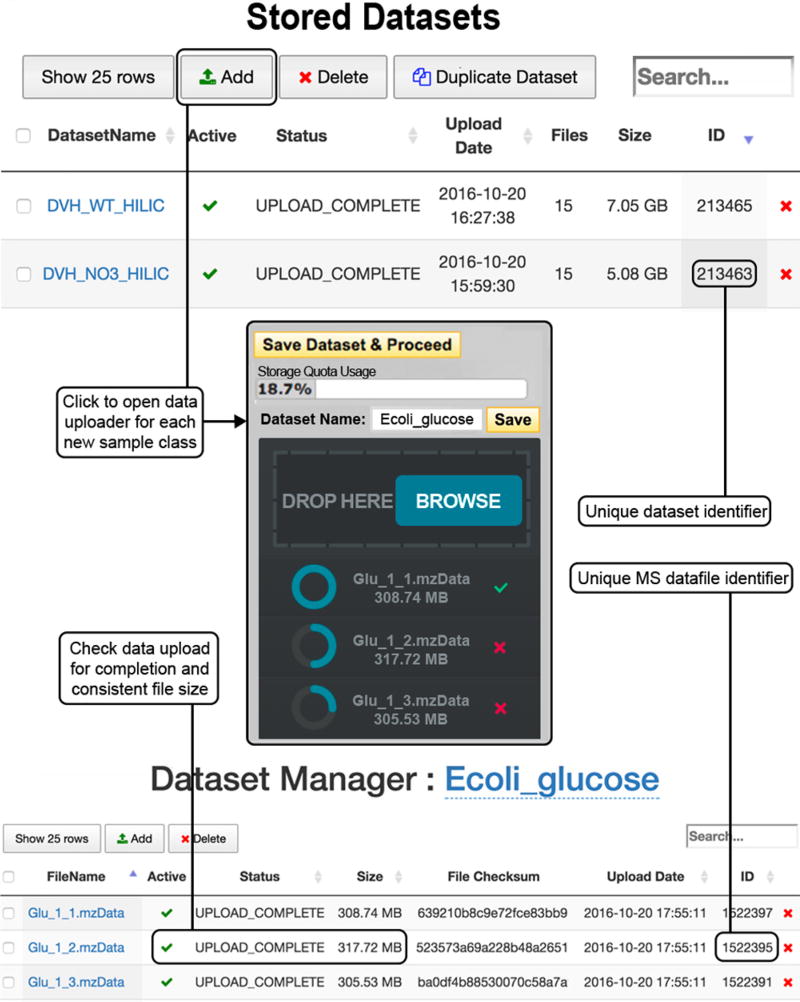
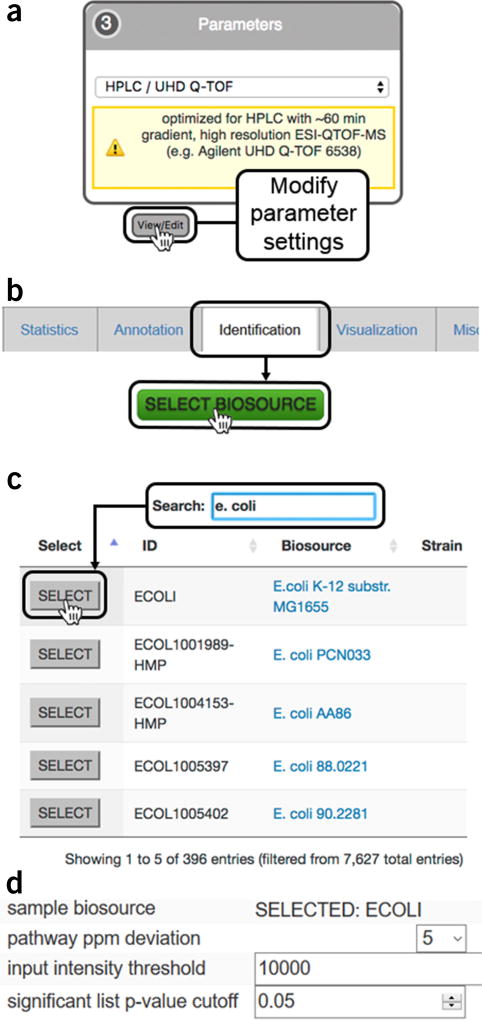
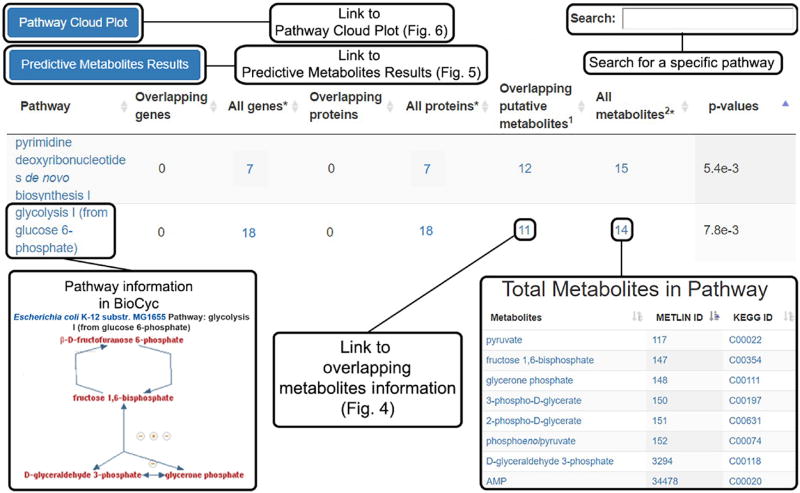
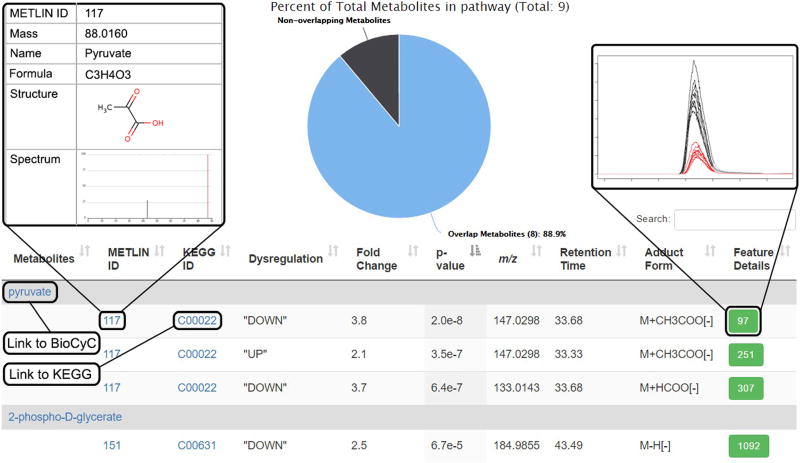
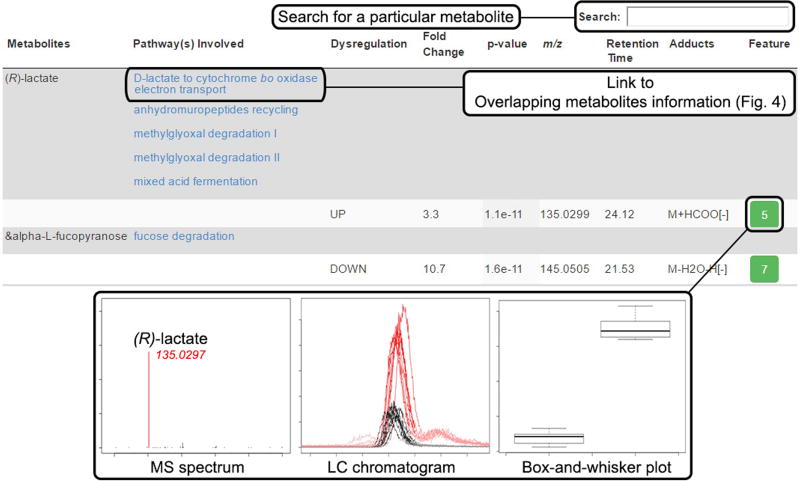
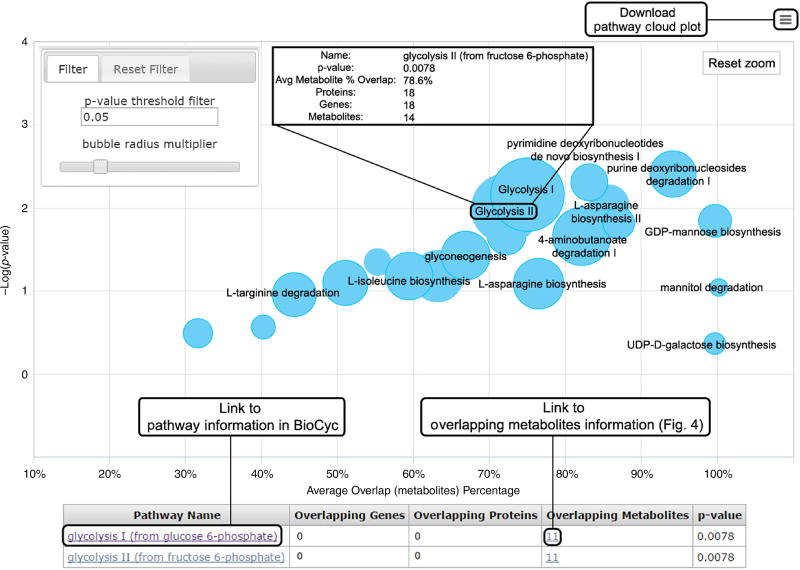
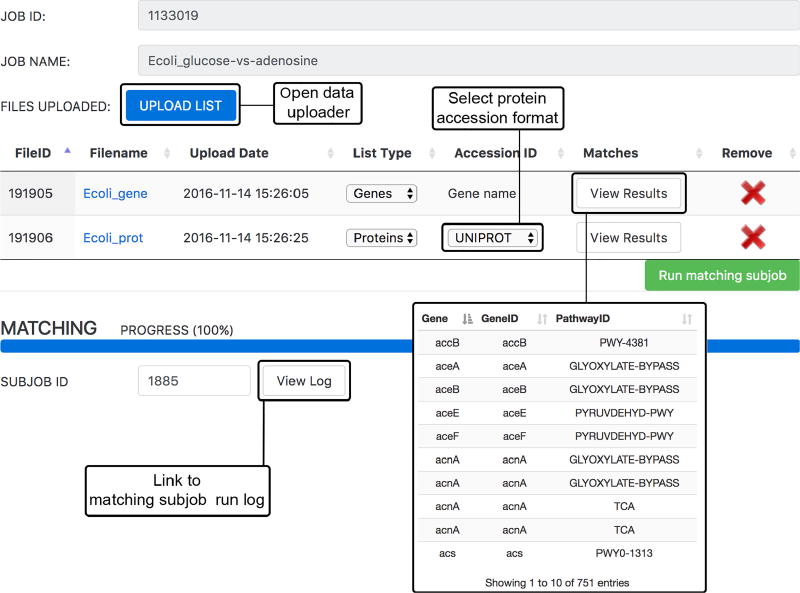
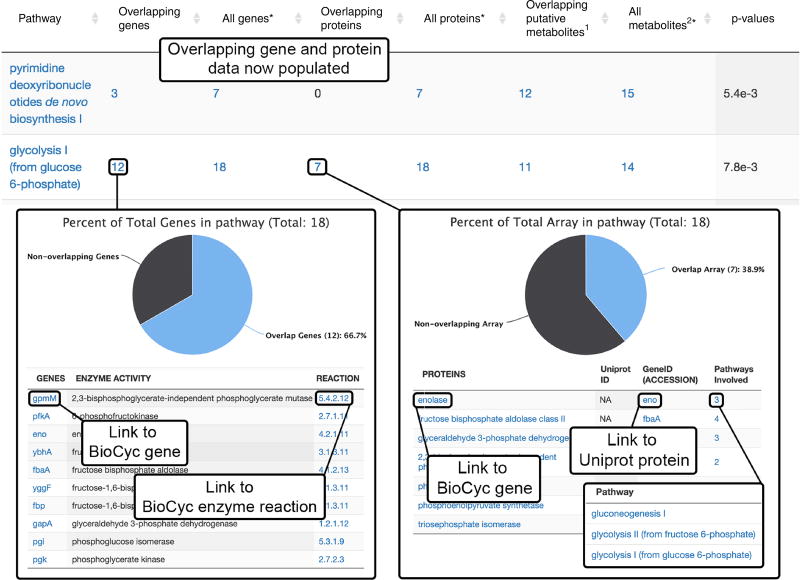
Comment in
-
Systems biology guided by XCMS Online metabolomics.Nat Methods. 2017 Apr 27;14(5):461-462. doi: 10.1038/nmeth.4260. Nat Methods. 2017. PMID: 28448069 Free PMC article. No abstract available.
References
-
- Goodacre R, Vaidyanathan S, Dunn WB, Harrigan GG, Kell DB. Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol. 2004;22:245–252. - PubMed
-
- Fondi M, Liò P. Multi-omics and metabolic modelling pipelines: challenges and tools for systems microbiology. Microbiol. Res. 2015;171:52–64. - PubMed
-
- Zampieri M, Sekar K, Zamboni N, Sauer U. Frontiers of high-throughput metabolomics. Curr. Opin. Chem. Biol. 2017;36:15–23. - PubMed
-
- Cajka T, Fiehn O. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Anal. Chem. 2016;88:524–545. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
