

Loss of BID Delays FASL-Induced Cell Death of Mouse Neutrophils and Aggravates DSS-Induced Weight Loss
- PMID: 29495595
- PMCID: PMC5877545
- DOI: 10.3390/ijms19030684
Loss of BID Delays FASL-Induced Cell Death of Mouse Neutrophils and Aggravates DSS-Induced Weight Loss
Abstract
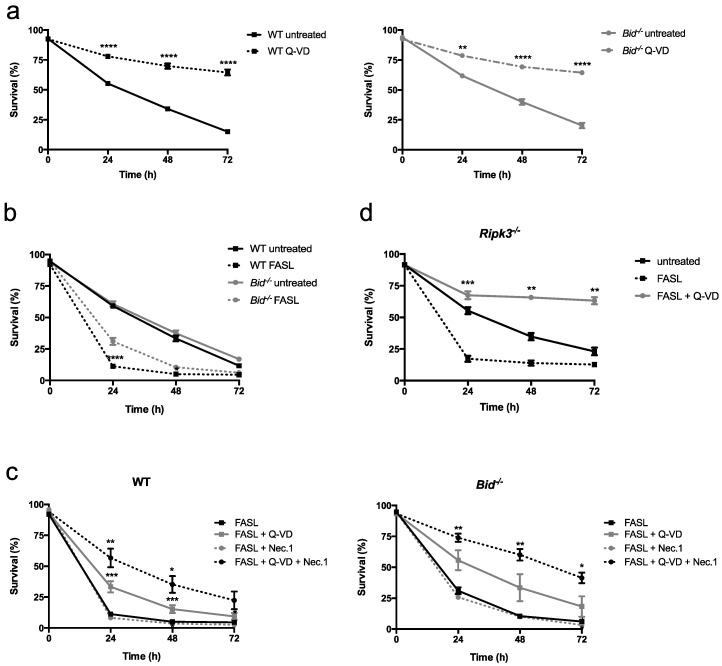
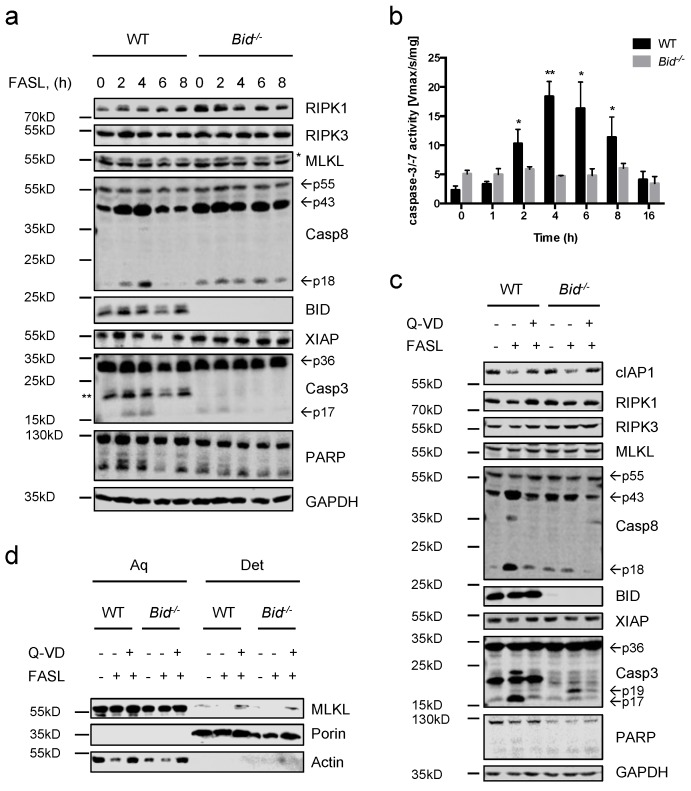
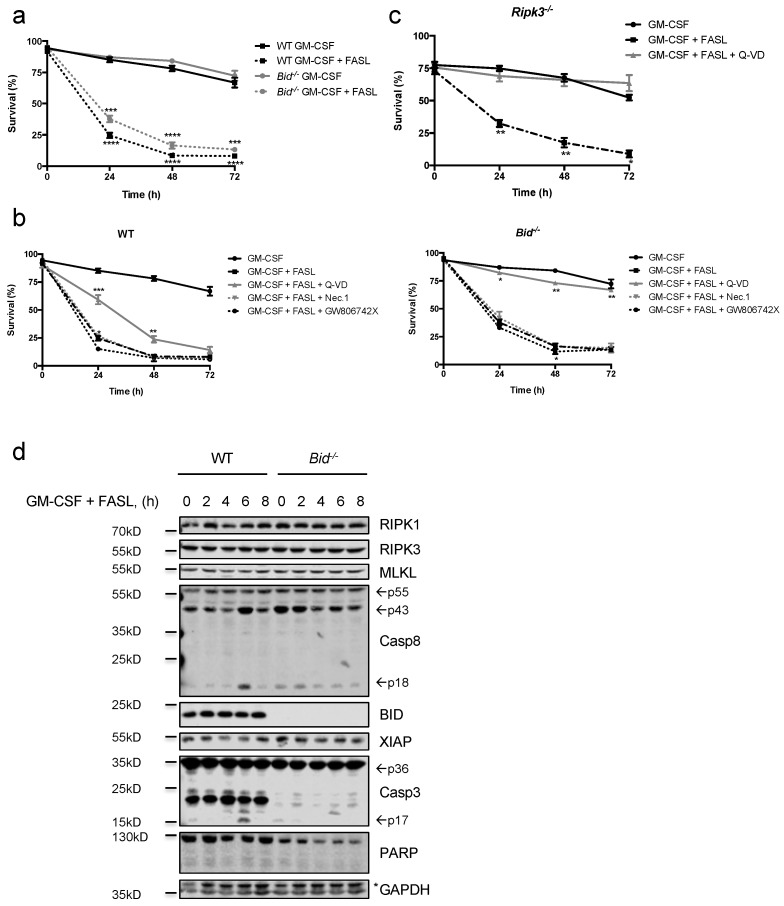
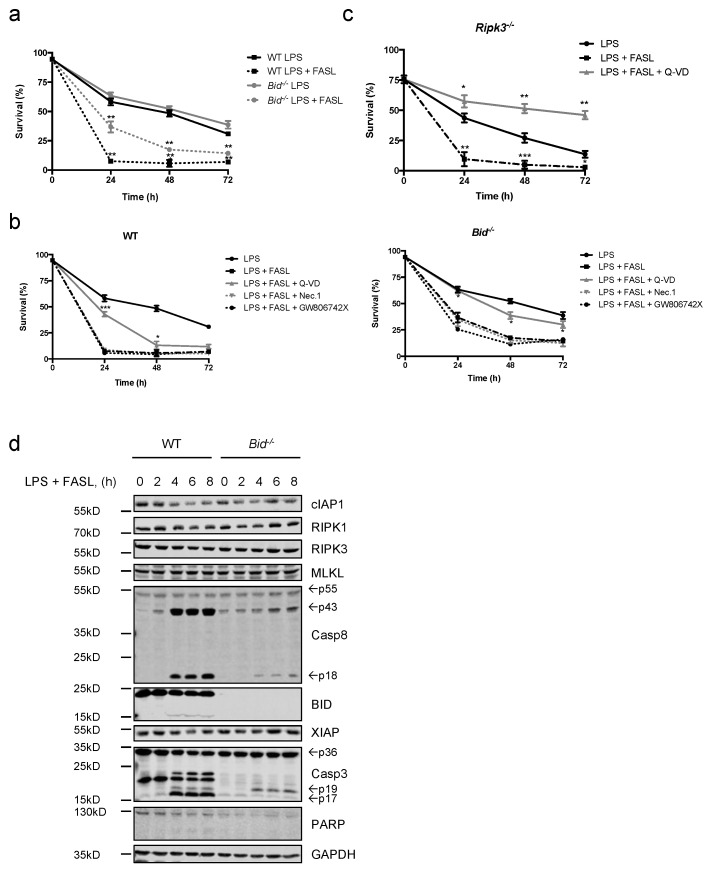
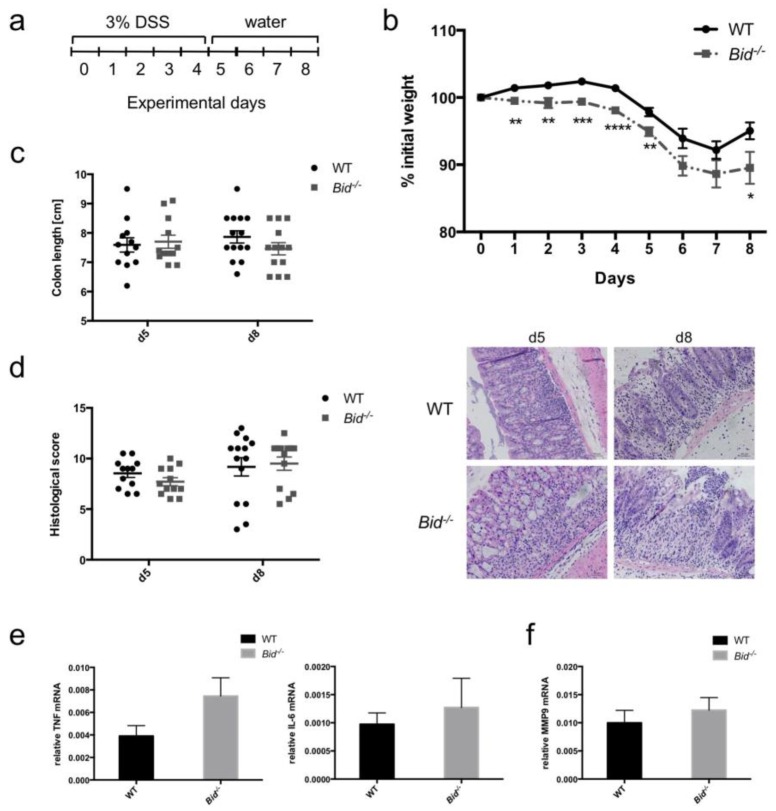
Neutrophils are key players in the early defense against invading pathogens. Due to their potent effector functions, programmed cell death of activated neutrophils has to be tightly controlled; however, its underlying mechanisms remain unclear. Fas ligand (FASL/CD95L) has been shown to induce neutrophil apoptosis, which is accelerated by the processing of the BH3-only protein BH3 interacting domain death agonist (BID) to trigger mitochondrial apoptotic events, and been attributed a regulatory role during viral and bacterial infections. Here, we show that, in accordance with previous works, mouse neutrophils underwent caspase-dependent apoptosis in response to FASL, and that this cell death was significantly delayed upon loss of BID. However, pan-caspase inhibition failed to protect mouse neutrophils from FASL-induced apoptosis and caused a switch to RIPK3-dependent necroptotic cell death. Intriguingly, such a switch was less evident in the absence of BID, particularly under inflammatory conditions. Delayed neutrophil apoptosis has been implicated in several auto-inflammatory diseases, including inflammatory bowel disease. We show that neutrophil and macrophage driven acute dextran sulfate sodium (DSS) induced colitis was slightly more aggravated in BID-deficient mice, based on significantly increased weight loss compared to wild-type controls. Taken together, our data support a central role for FASL > FAS and BID in mouse neutrophil cell death and further underline the anti-inflammatory role of BID.
Keywords: BID; FAS/CD95; RIPK3; apoptosis; caspases; colitis; inflammation; mouse model; necroptosis; neutrophil.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Colotta F., Re F., Polentarutti N., Sozzani S., Mantovani A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood. 1992;80:2012–2020. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
