

Unravelling the molecular basis for regulatory T-cell plasticity and loss of function in disease
- PMID: 29497530
- PMCID: PMC5827651
- DOI: 10.1002/cti2.1011
Unravelling the molecular basis for regulatory T-cell plasticity and loss of function in disease
Abstract
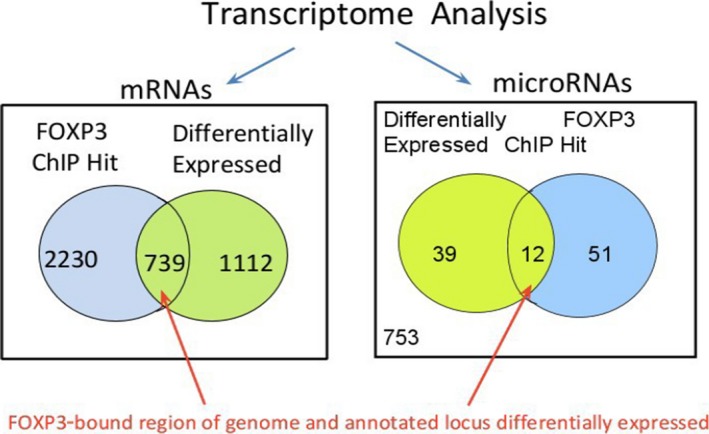
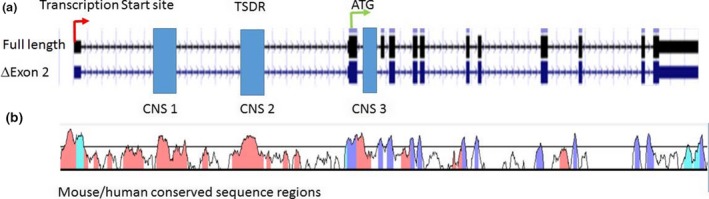
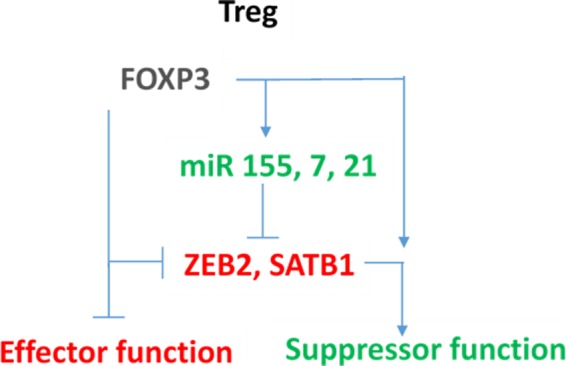
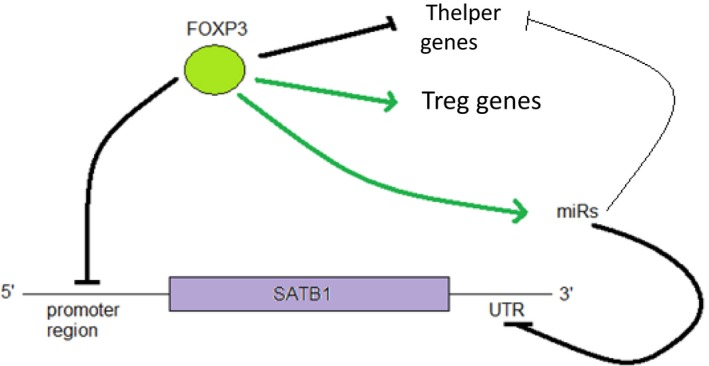
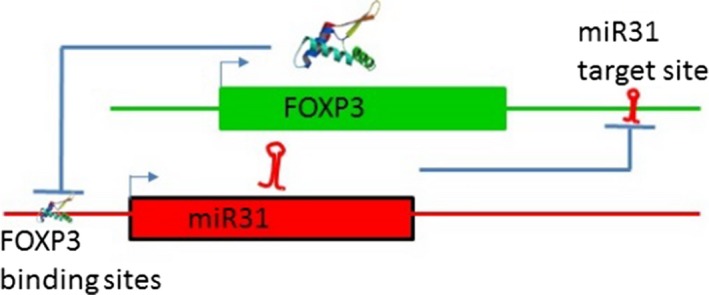
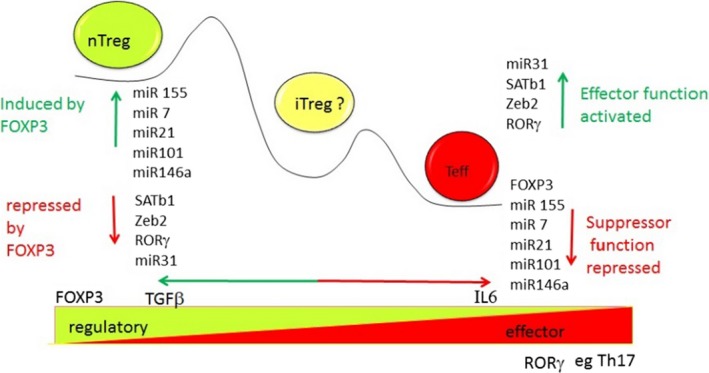
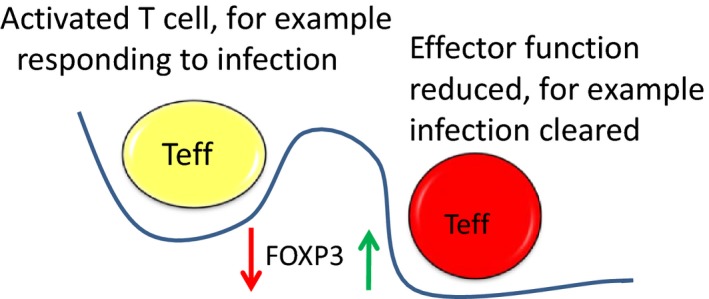
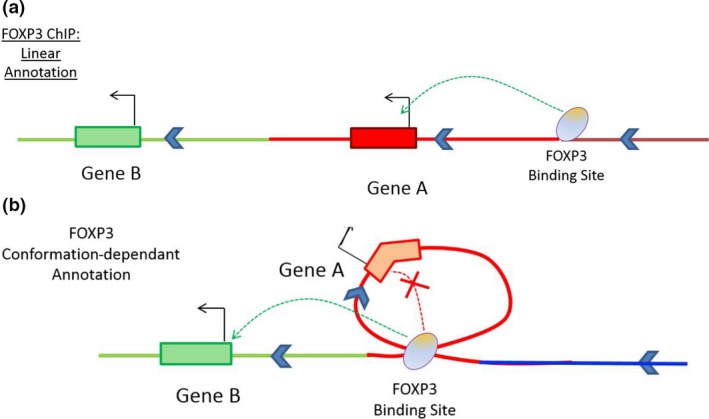
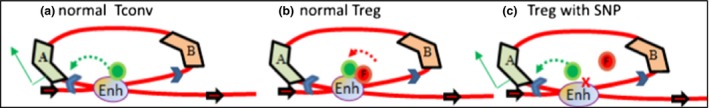
Regulatory T cells (Treg) are critical for preventing autoimmunity and curtailing responses of conventional effector T cells (Tconv). The reprogramming of T-cell fate and function to generate Treg requires switching on and off of key gene regulatory networks, which may be initiated by a subtle shift in expression levels of specific genes. This can be achieved by intermediary regulatory processes that include microRNA and long noncoding RNA-based regulation of gene expression. There are well-documented microRNA profiles in Treg and Tconv, and these can operate to either reinforce or reduce expression of a specific set of target genes, including FOXP3 itself. This type of feedforward/feedback regulatory loop is normally stable in the steady state, but can alter in response to local cues or genetic risk. This may go some way to explaining T-cell plasticity. In addition, in chronic inflammation or autoimmunity, altered Treg/Tconv function may be influenced by changes in enhancer-promoter interactions, which are highly cell type-specific. These interactions are impacted by genetic risk based on genome-wide association studies and may cause subtle alterations to the gene regulatory networks controlled by or controlling FOXP3 and its target genes. Recent insights into the 3D organisation of chromatin and the mapping of noncoding regulatory regions to the genes they control are shedding new light on the direct impact of genetic risk on T-cell function and susceptibility to inflammatory and autoimmune conditions.
Keywords: Treg FOXP3; T‐cell fate; T‐cell plasticity; gene regulation; genetic risk of disease; microRNA.
Figures
References
-
- Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non‐inflammatory conditions. Semin Immunol 2011; 23: 424–430. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources