

Factors influencing readthrough therapy for frequent cystic fibrosis premature termination codons
- PMID: 29497617
- PMCID: PMC5827411
- DOI: 10.1183/23120541.00080-2017
Factors influencing readthrough therapy for frequent cystic fibrosis premature termination codons
Erratum in
-
Erratum: "Factors influencing readthrough therapy for frequent cystic fibrosis premature termination codons" Iwona Pranke, Laure Bidou, Natacha Martin, Sandra Blanchet, Aurélie Hatton, Sabrina Karri, David Cornu, Bruno Costes, Benoit Chevalier, Danielle Tondelier, Emmanuelle Girodon, Matthieu Coupet, Aleksander Edelman, Pascale Fanen, Olivier Namy, Isabelle Sermet-Gaudelus and Alexandre Hinzpeter.ERJ Open Res 2018; 4: 00080-2017.ERJ Open Res. 2018 Jul 13;4(3):00080-2017-AUT. doi: 10.1183/23120541.50080-2017. eCollection 2018 Jul. ERJ Open Res. 2018. PMID: 30035115 Free PMC article.
Abstract
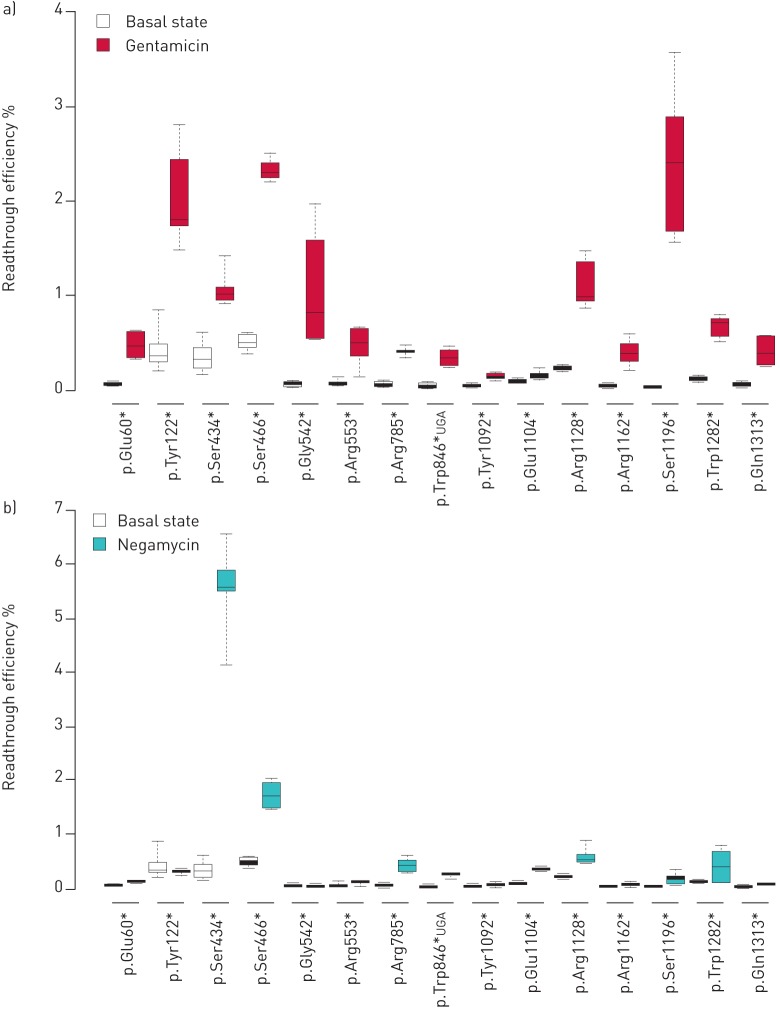
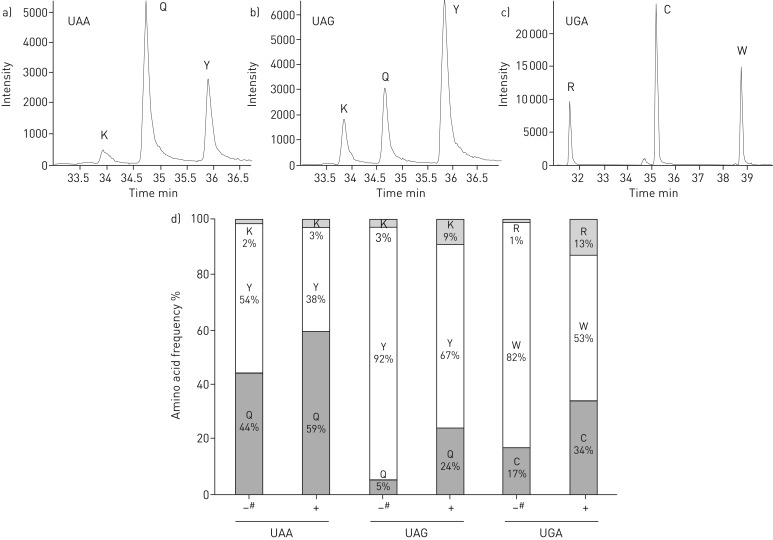
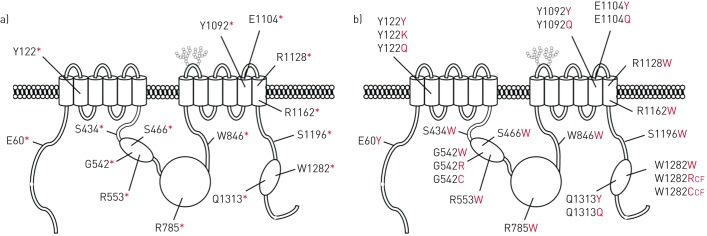
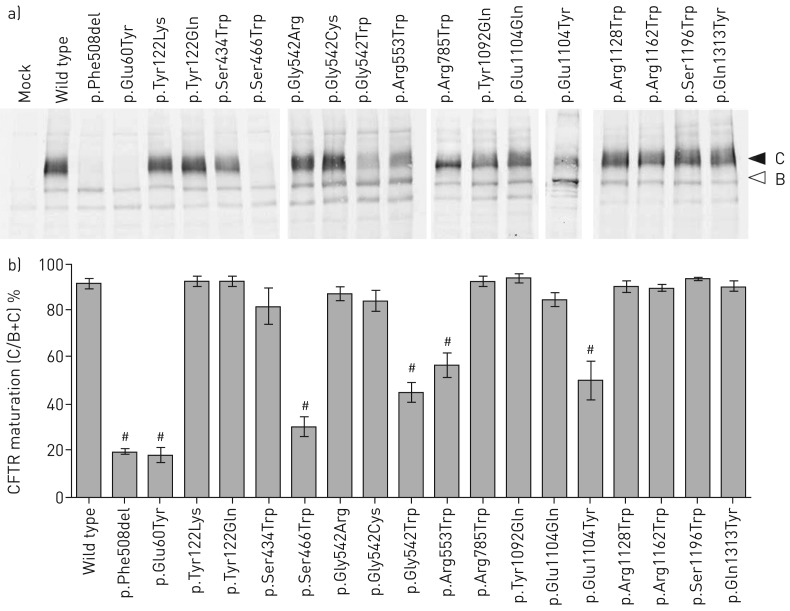
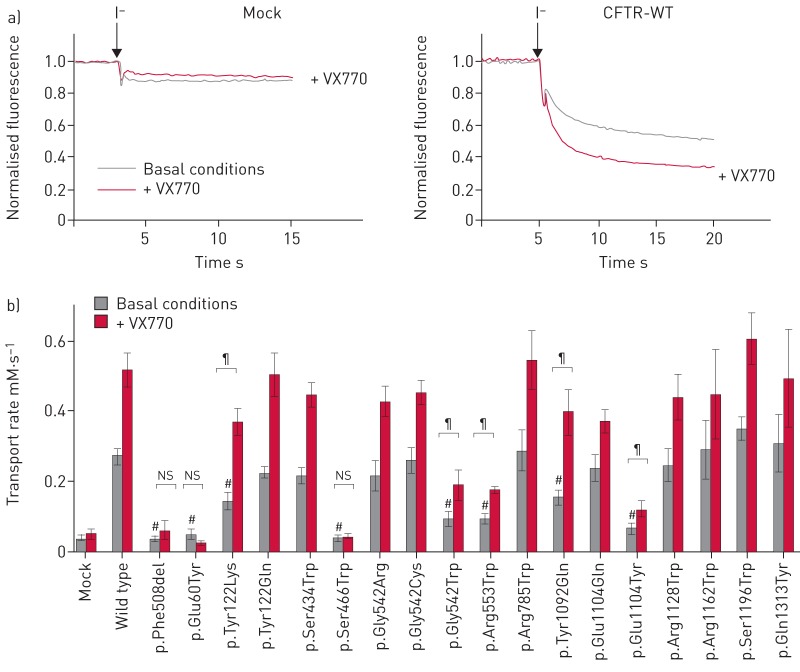
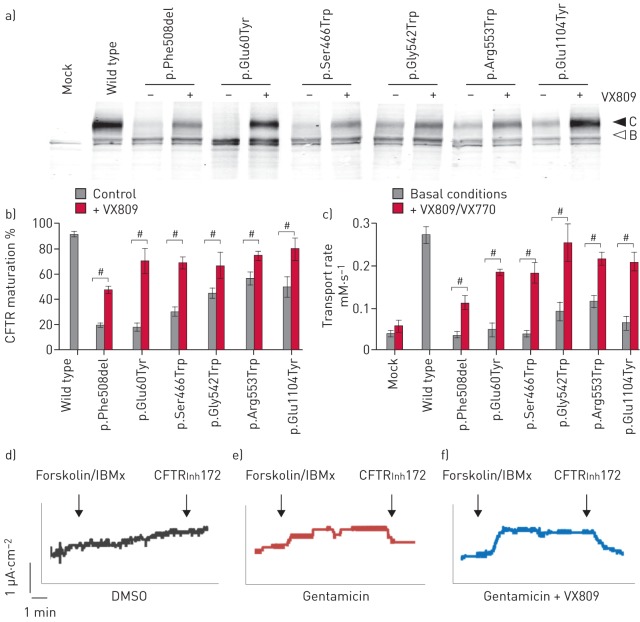
Premature termination codons (PTCs) are generally associated with severe forms of genetic diseases. Readthrough of in-frame PTCs using small molecules is a promising therapeutic approach. Nonetheless, the outcome of preclinical studies has been low and variable. Treatment efficacy depends on: 1) the level of drug-induced readthrough, 2) the amount of target transcripts, and 3) the activity of the recoded protein. The aim of the present study was to identify, in the cystic fibrosis transmembrane conductance regulator (CFTR) model, recoded channels from readthrough therapy that may be enhanced using CFTR modulators. First, drug-induced readthrough of 15 PTCs was measured using a dual reporter system under basal conditions and in response to gentamicin and negamycin. Secondly, exon skipping associated with these PTCs was evaluated with a minigene system. Finally, incorporated amino acids were identified by mass spectrometry and the function of the predicted recoded CFTR channels corresponding to these 15 PTCs was measured. Nonfunctional channels were subjected to CFTR-directed ivacaftor-lumacaftor treatments. The results demonstrated that CFTR modulators increased activity of recoded channels, which could also be confirmed in cells derived from a patient. In conclusion, this work will provide a framework to adapt treatments to the patient's genotype by identifying the most efficient molecule for each PTC and the recoded channels needing co-therapies to rescue channel function.
Conflict of interest statement
Conflict of interest: None declared.
Figures

References
LinkOut - more resources
Full Text Sources
Other Literature Sources