

Mutations in ATP1A1 Cause Dominant Charcot-Marie-Tooth Type 2
- PMID: 29499166
- PMCID: PMC5985288
- DOI: 10.1016/j.ajhg.2018.01.023
Mutations in ATP1A1 Cause Dominant Charcot-Marie-Tooth Type 2
Abstract
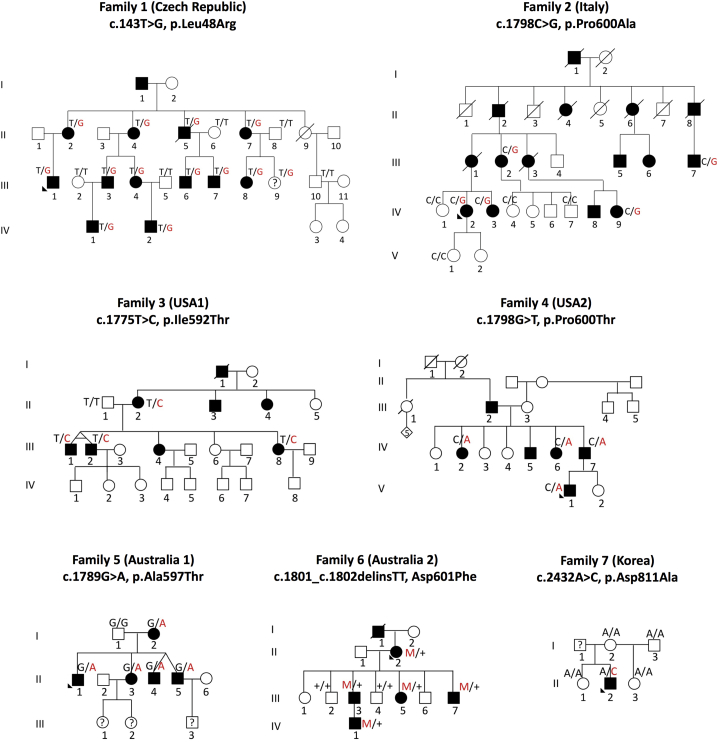
Although mutations in more than 90 genes are known to cause CMT, the underlying genetic cause of CMT remains unknown in more than 50% of affected individuals. The discovery of additional genes that harbor CMT2-causing mutations increasingly depends on sharing sequence data on a global level. In this way-by combining data from seven countries on four continents-we were able to define mutations in ATP1A1, which encodes the alpha1 subunit of the Na+,K+-ATPase, as a cause of autosomal-dominant CMT2. Seven missense changes were identified that segregated within individual pedigrees: c.143T>G (p.Leu48Arg), c.1775T>C (p.Ile592Thr), c.1789G>A (p.Ala597Thr), c.1801_1802delinsTT (p.Asp601Phe), c.1798C>G (p.Pro600Ala), c.1798C>A (p.Pro600Thr), and c.2432A>C (p.Asp811Ala). Immunostaining peripheral nerve axons localized ATP1A1 to the axolemma of myelinated sensory and motor axons and to Schmidt-Lanterman incisures of myelin sheaths. Two-electrode voltage clamp measurements on Xenopus oocytes demonstrated significant reduction in Na+ current activity in some, but not all, ouabain-insensitive ATP1A1 mutants, suggesting a loss-of-function defect of the Na+,K+ pump. Five mutants fall into a remarkably narrow motif within the helical linker region that couples the nucleotide-binding and phosphorylation domains. These findings identify a CMT pathway and a potential target for therapy development in degenerative diseases of peripheral nerve axons.
Keywords: ATP1A1; CMT; Charcot-Marie-Tooth; Mendelian disease; Na(+),K(+) ATPase; axonal neuropathy; genetic matchmaking.
Copyright © 2018 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures


References
-
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974;6:98–118. - PubMed
-
- Pareyson D., Saveri P., Pisciotta C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr. Opin. Neurol. 2017;30:471–480. - PubMed
-
- Fridman V., Bundy B., Reilly M.M., Pareyson D., Bacon C., Burns J., Day J., Feely S., Finkel R.S., Grider T., Inherited Neuropathies Consortium CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J. Neurol. Neurosurg. Psychiatry. 2015;86:873–878. - PMC - PubMed
-
- Rossor A.M., Polke J.M., Houlden H., Reilly M.M. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat. Rev. Neurol. 2013;9:562–571. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
