

Complete avian malaria parasite genomes reveal features associated with lineage-specific evolution in birds and mammals
- PMID: 29500236
- PMCID: PMC5880244
- DOI: 10.1101/gr.218123.116
Complete avian malaria parasite genomes reveal features associated with lineage-specific evolution in birds and mammals
Abstract
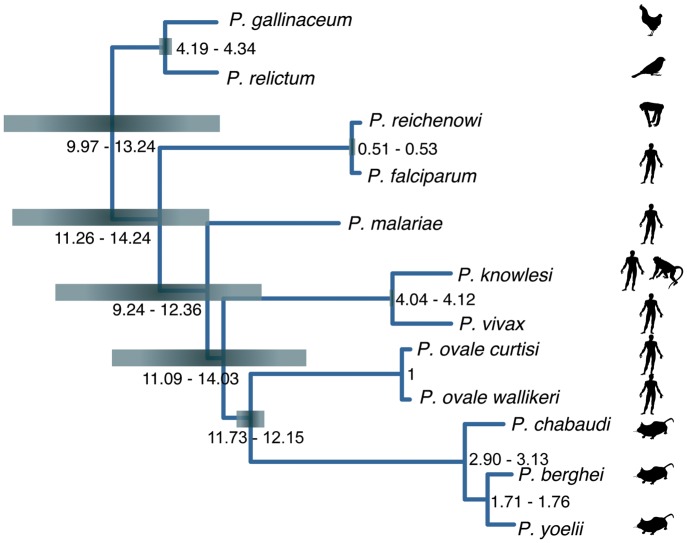
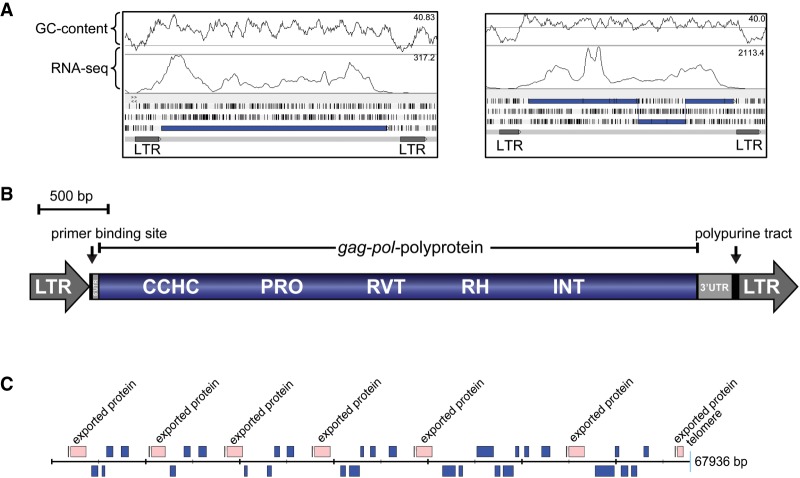
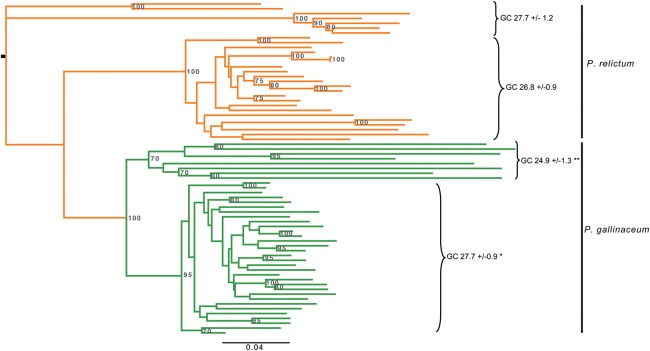
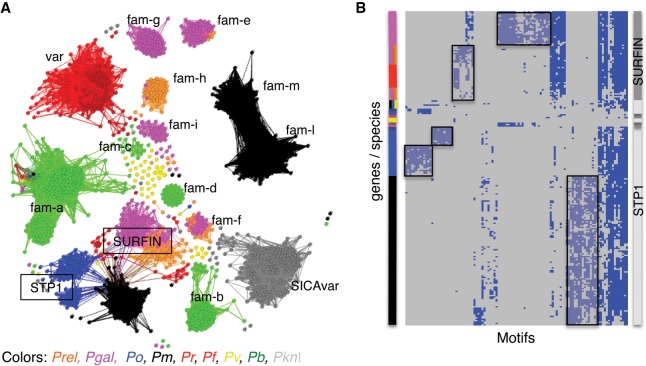
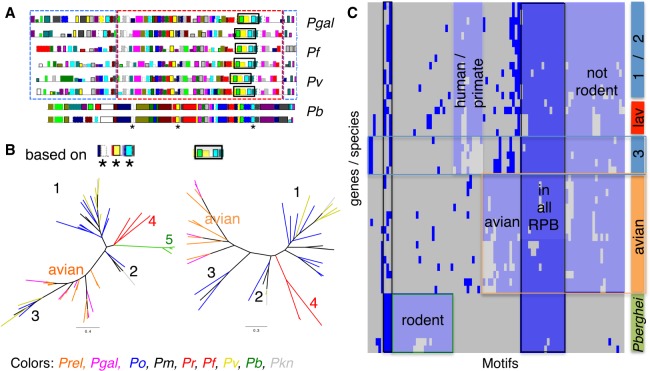
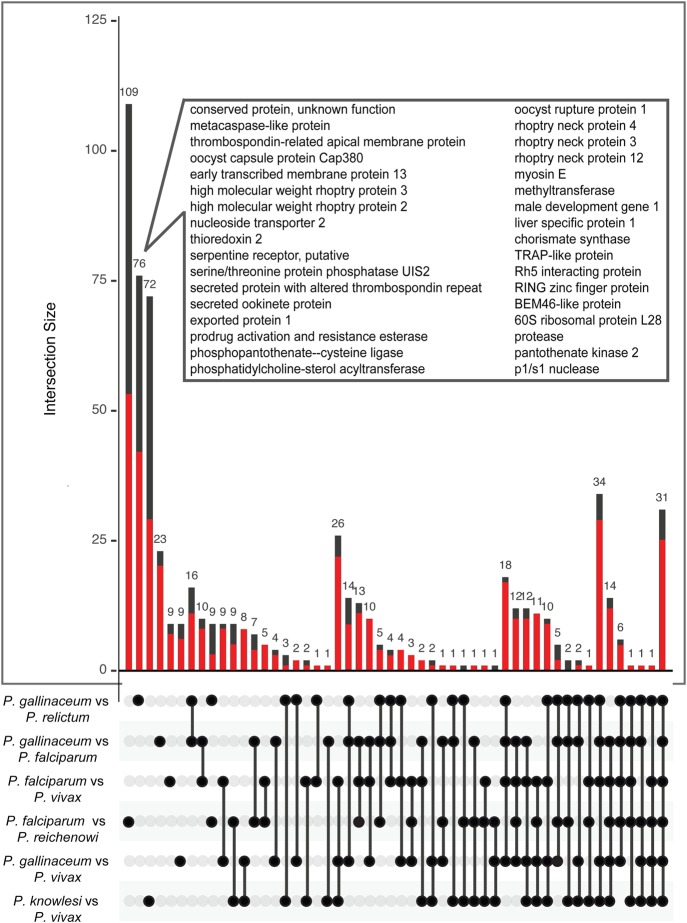
Avian malaria parasites are prevalent around the world and infect a wide diversity of bird species. Here, we report the sequencing and analysis of high-quality draft genome sequences for two avian malaria species, Plasmodium relictum and Plasmodium gallinaceum We identify 50 genes that are specific to avian malaria, located in an otherwise conserved core of the genome that shares gene synteny with all other sequenced malaria genomes. Phylogenetic analysis suggests that the avian malaria species form an outgroup to the mammalian Plasmodium species, and using amino acid divergence between species, we estimate the avian- and mammalian-infective lineages diverged in the order of 10 million years ago. Consistent with their phylogenetic position, we identify orthologs of genes that had previously appeared to be restricted to the clades of parasites containing Plasmodium falciparum and Plasmodium vivax, the species with the greatest impact on human health. From these orthologs, we explore differential diversifying selection across the genus and show that the avian lineage is remarkable in the extent to which invasion-related genes are evolving. The subtelomeres of the P. relictum and P. gallinaceum genomes contain several novel gene families, including an expanded surf multigene family. We also identify an expansion of reticulocyte binding protein homologs in P. relictum, and within these proteins, we detect distinct regions that are specific to nonhuman primate, humans, rodent, and avian hosts. For the first time in the Plasmodium lineage, we find evidence of transposable elements, including several hundred fragments of LTR-retrotransposons in both species and an apparently complete LTR-retrotransposon in the genome of P. gallinaceum.
© 2018 Böhme et al.; Published by Cold Spring Harbor Laboratory Press.
Figures

References
-
- Asghar M, Hasselquist D, Hansson B, Zehtindjiev P, Westerdahl H, Bensch S. 2015. Chronic infection. Hidden costs of infection: Chronic malaria accelerates telomere degradation and senescence in wild birds. Science 347: 436–438. - PubMed
-
- Atkinson CT, Dusek RJ, Woods KL, Iko WM. 2000. Pathogenicity of avian malaria in experimentally-infected Hawaii Amakihi. J Wildl Dis 36: 197–204. - PubMed
-
- Bensch S, Hellgren O, Pérez-Tris J. 2009. MalAvi: a public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Mol Ecol Resour 9: 1353–1358. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases