

Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits
- PMID: 29500431
- PMCID: PMC5834629
- DOI: 10.1038/s41467-018-03371-0
Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits
Abstract
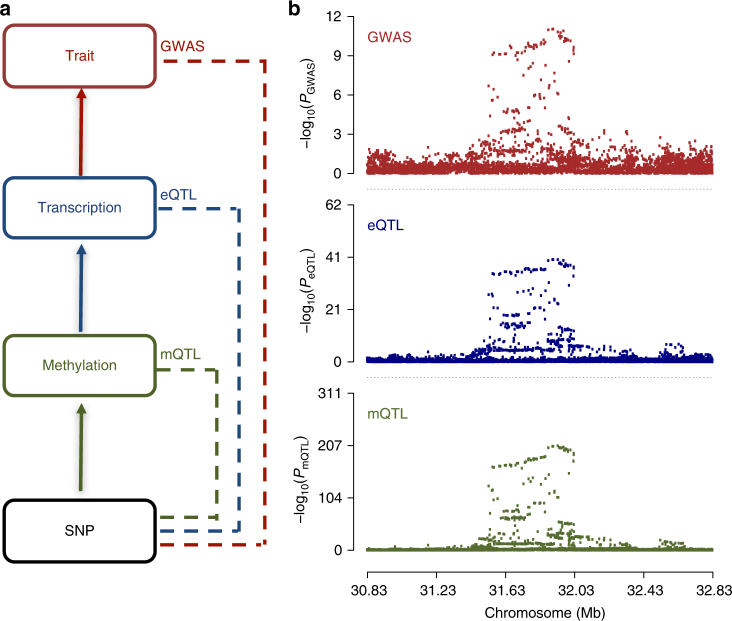
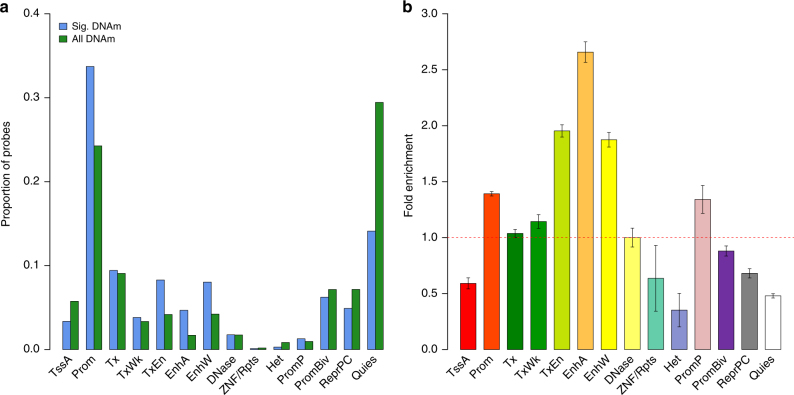
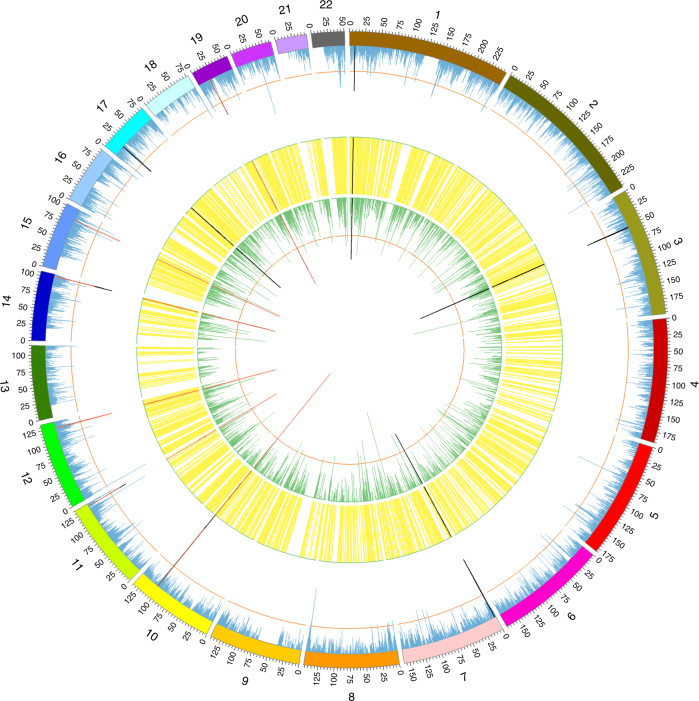
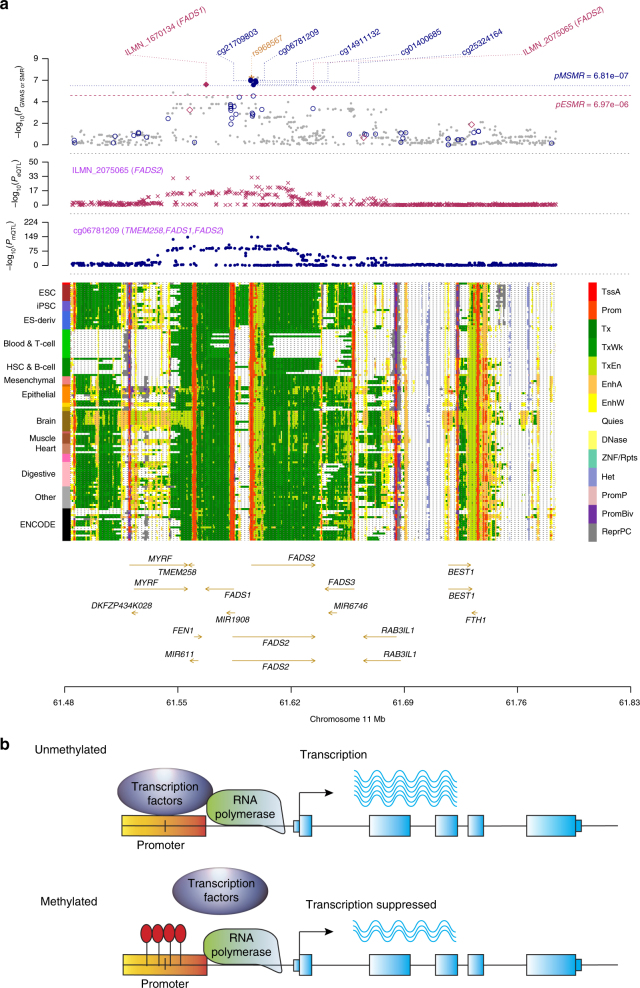
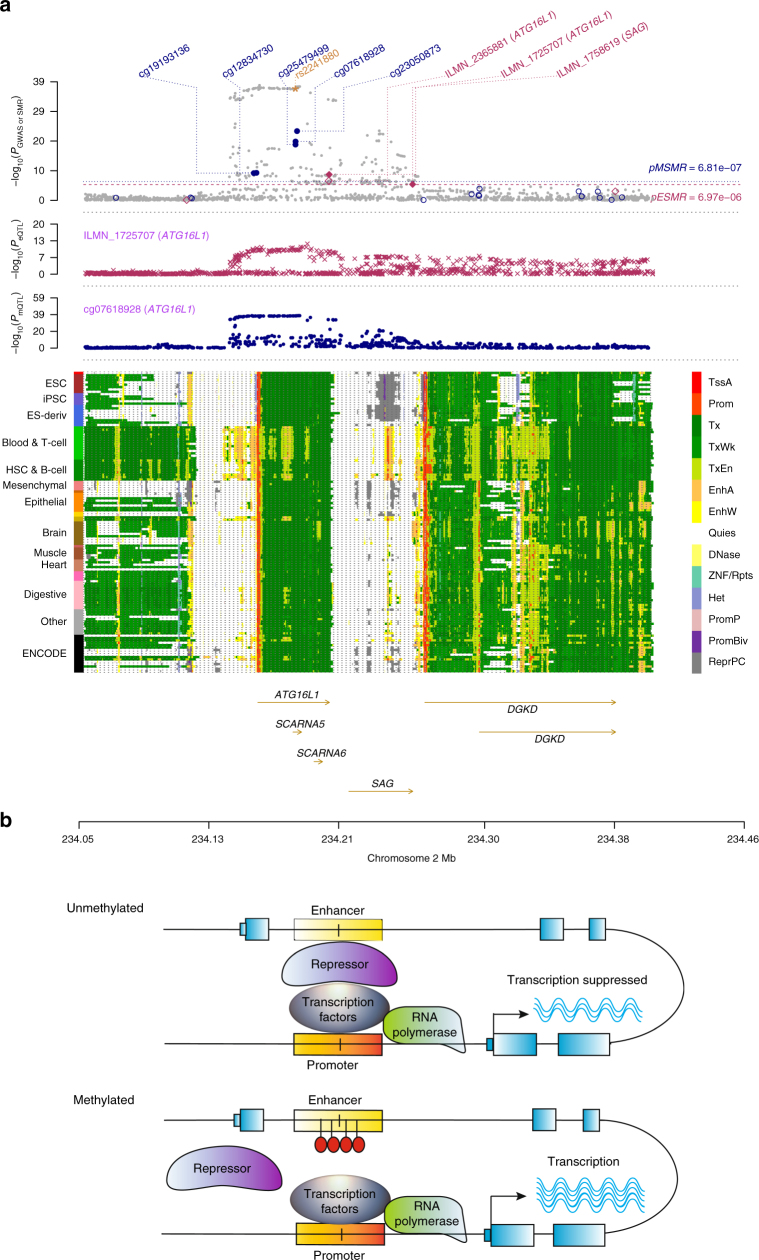
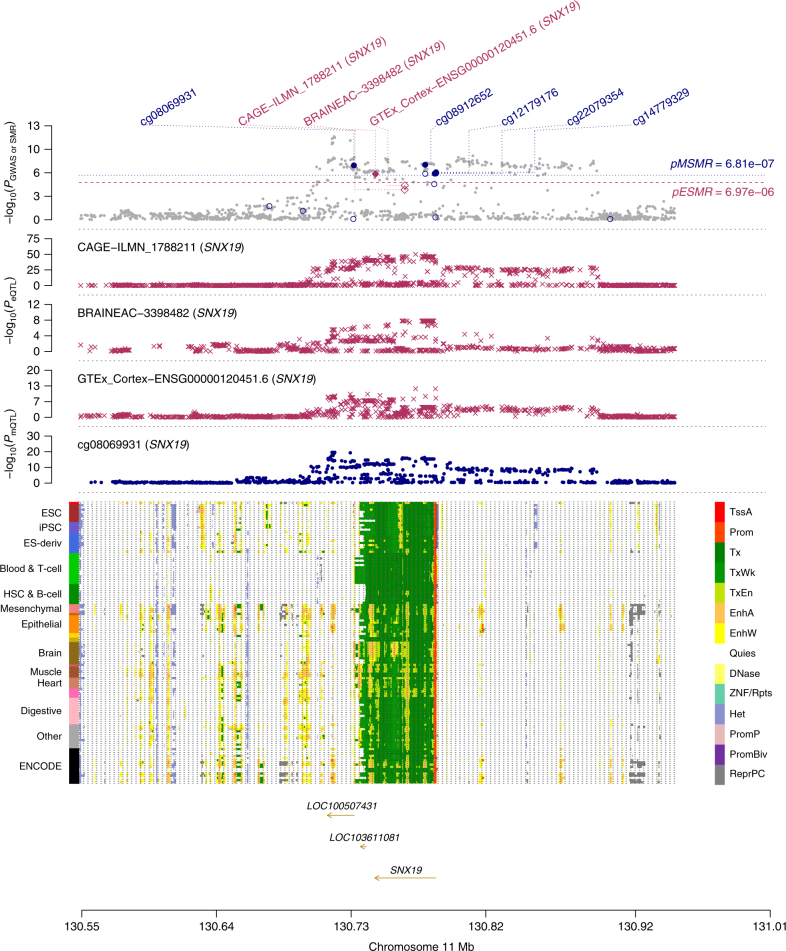
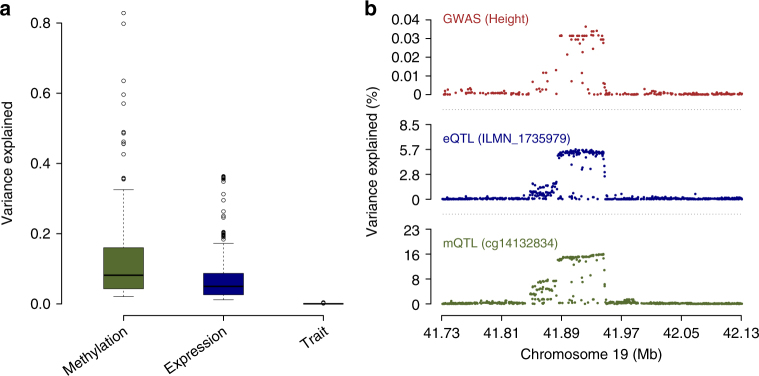
The identification of genes and regulatory elements underlying the associations discovered by GWAS is essential to understanding the aetiology of complex traits (including diseases). Here, we demonstrate an analytical paradigm of prioritizing genes and regulatory elements at GWAS loci for follow-up functional studies. We perform an integrative analysis that uses summary-level SNP data from multi-omics studies to detect DNA methylation (DNAm) sites associated with gene expression and phenotype through shared genetic effects (i.e., pleiotropy). We identify pleiotropic associations between 7858 DNAm sites and 2733 genes. These DNAm sites are enriched in enhancers and promoters, and >40% of them are mapped to distal genes. Further pleiotropic association analyses, which link both the methylome and transcriptome to 12 complex traits, identify 149 DNAm sites and 66 genes, indicating a plausible mechanism whereby the effect of a genetic variant on phenotype is mediated by genetic regulation of transcription through DNAm.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
