

Cardiomyocyte hypertrophy induced by Endonuclease G deficiency requires reactive oxygen radicals accumulation and is inhibitable by the micropeptide humanin
- PMID: 29502044
- PMCID: PMC5952880
- DOI: 10.1016/j.redox.2018.02.021
Cardiomyocyte hypertrophy induced by Endonuclease G deficiency requires reactive oxygen radicals accumulation and is inhibitable by the micropeptide humanin
Abstract
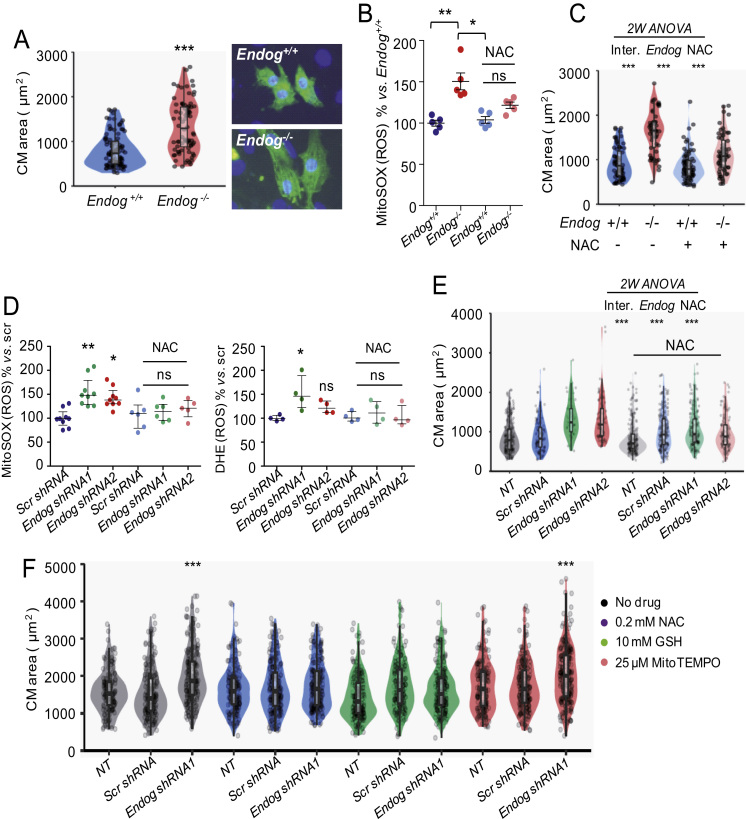
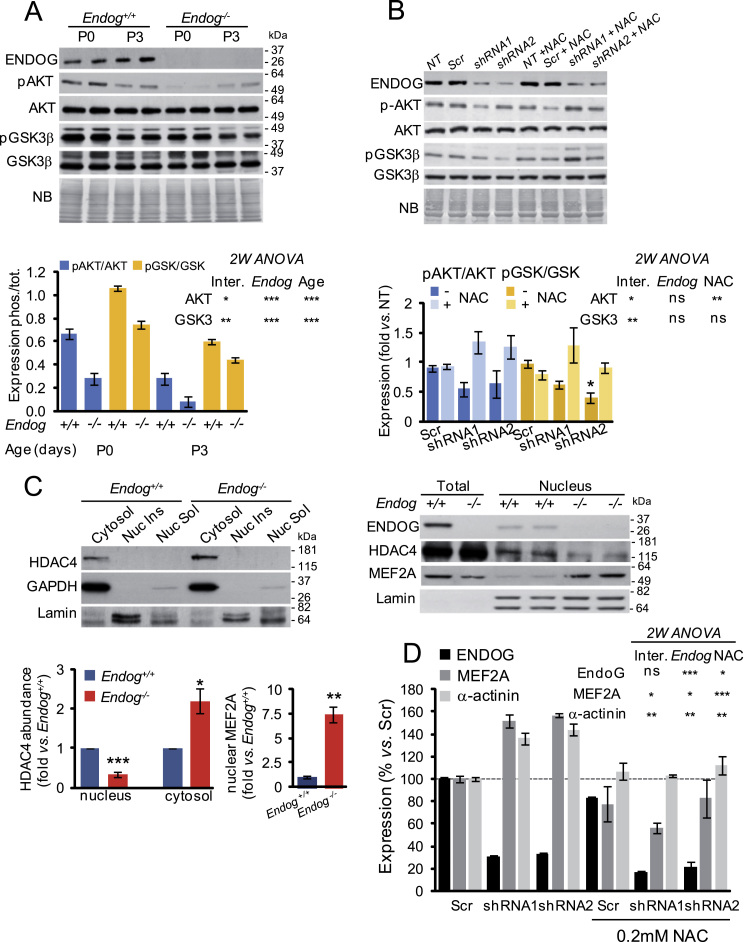
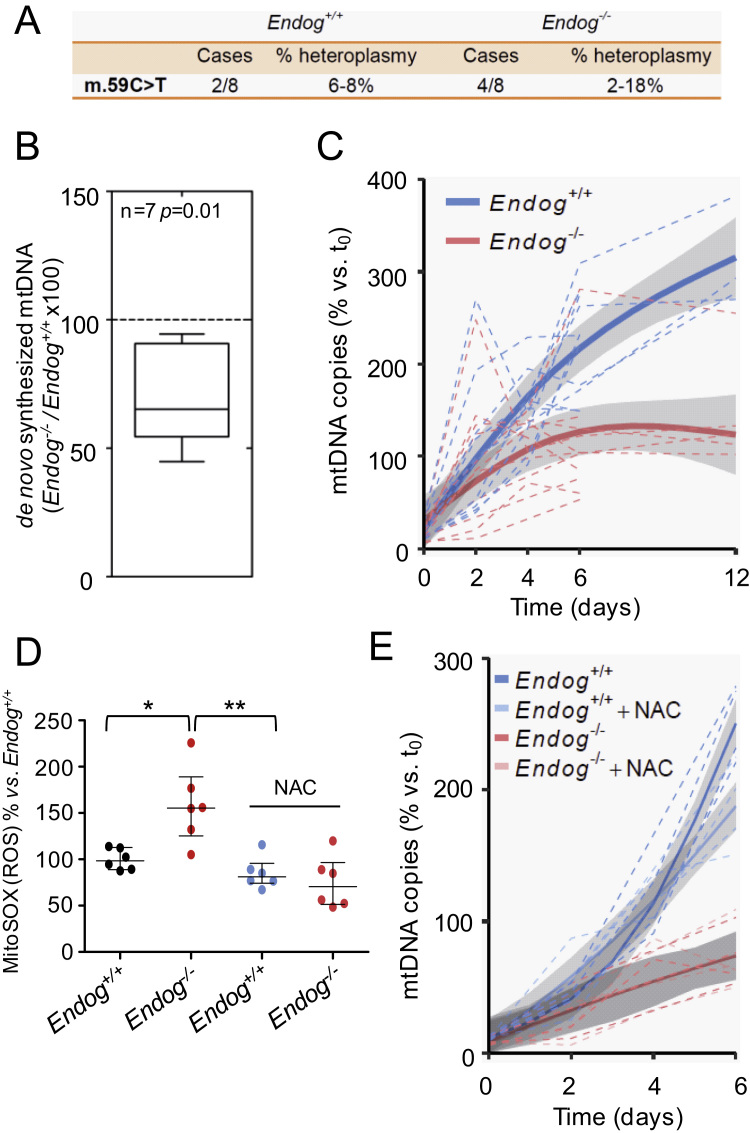
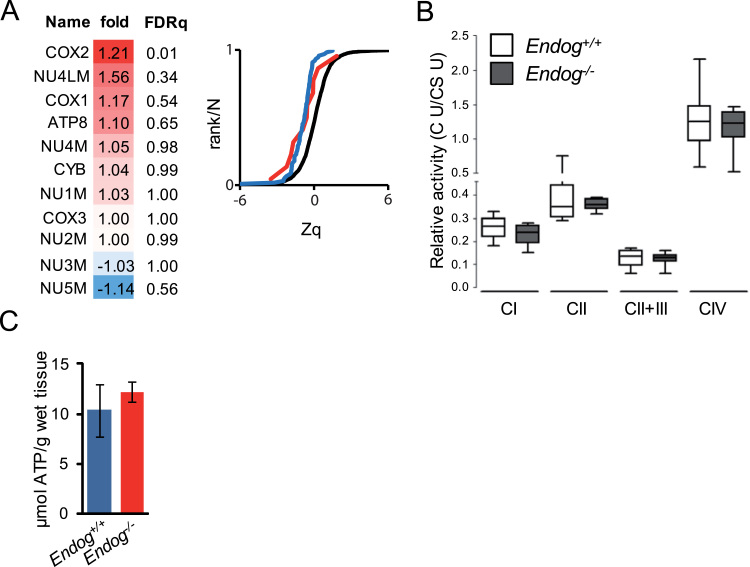
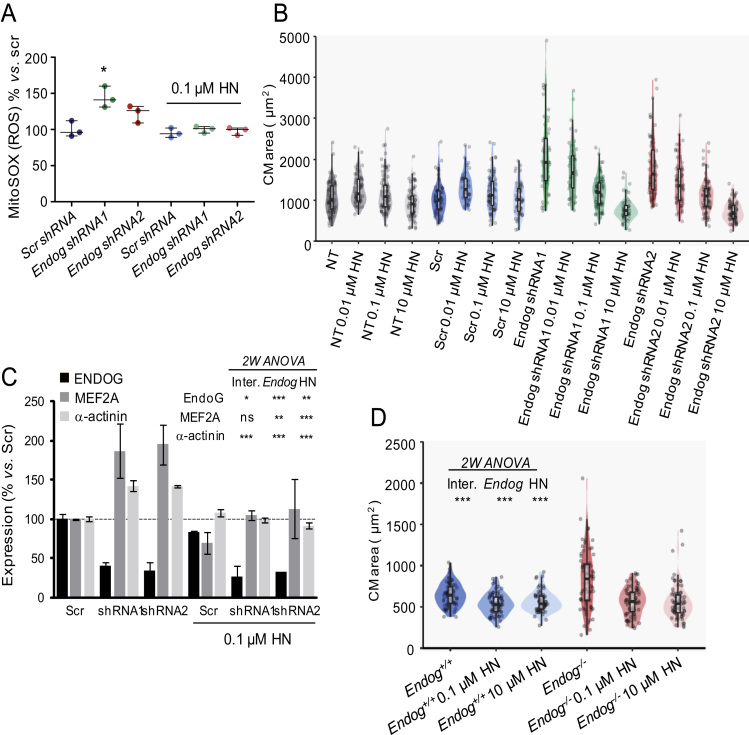
The endonuclease G gene (Endog), which codes for a mitochondrial nuclease, was identified as a determinant of cardiac hypertrophy. How ENDOG controls cardiomyocyte growth is still unknown. Thus, we aimed at finding the link between ENDOG activity and cardiomyocyte growth. Endog deficiency induced reactive oxygen species (ROS) accumulation and abnormal growth in neonatal rodent cardiomyocytes, altering the AKT-GSK3β and Class-II histone deacethylases (HDAC) signal transduction pathways. These effects were blocked by ROS scavengers. Lack of ENDOG reduced mitochondrial DNA (mtDNA) replication independently of ROS accumulation. Because mtDNA encodes several subunits of the mitochondrial electron transport chain, whose activity is an important source of cellular ROS, we investigated whether Endog deficiency compromised the expression and activity of the respiratory chain complexes but found no changes in these parameters nor in ATP content. MtDNA also codes for humanin, a micropeptide with possible metabolic functions. Nanomolar concentrations of synthetic humanin restored normal ROS levels and cell size in Endog-deficient cardiomyocytes. These results support the involvement of redox signaling in the control of cardiomyocyte growth by ENDOG and suggest a pathway relating mtDNA content to the regulation of cell growth probably involving humanin, which prevents reactive oxygen radicals accumulation and hypertrophy induced by Endog deficiency.
Keywords: ENDOG; cardiac hypertrophy; humanin; mitochondrial DNA.
Copyright © 2018 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
Similar articles
-
Involvement of the mitochondrial nuclease EndoG in the regulation of cell proliferation through the control of reactive oxygen species.Redox Biol. 2020 Oct;37:101736. doi: 10.1016/j.redox.2020.101736. Epub 2020 Sep 24. Redox Biol. 2020. PMID: 33032073 Free PMC article.
-
Endonuclease G is a novel determinant of cardiac hypertrophy and mitochondrial function.Nature. 2011 Oct 5;478(7367):114-8. doi: 10.1038/nature10490. Nature. 2011. PMID: 21979051 Free PMC article.
-
Loss of mitochondrial exo/endonuclease EXOG affects mitochondrial respiration and induces ROS-mediated cardiomyocyte hypertrophy.Am J Physiol Cell Physiol. 2015 Jan 15;308(2):C155-63. doi: 10.1152/ajpcell.00227.2014. Epub 2014 Nov 5. Am J Physiol Cell Physiol. 2015. PMID: 25377088
-
Mitochondrial peptides-appropriate options for therapeutic exploitation.Cell Tissue Res. 2019 Aug;377(2):161-165. doi: 10.1007/s00441-019-03049-z. Epub 2019 May 27. Cell Tissue Res. 2019. PMID: 31131430 Review.
-
ROS signalling between endothelial cells and cardiac cells.Cardiovasc Res. 2014 May 1;102(2):249-57. doi: 10.1093/cvr/cvu050. Epub 2014 Mar 3. Cardiovasc Res. 2014. PMID: 24591150 Review.
Cited by
-
Analysis of Therapeutic Targets of A Novel Peptide Athycaltide-1 in the Treatment of Isoproterenol-Induced Pathological Myocardial Hypertrophy.Cardiovasc Ther. 2022 May 2;2022:2715084. doi: 10.1155/2022/2715084. eCollection 2022. Cardiovasc Ther. 2022. PMID: 35599721 Free PMC article.
-
ENDOG Impacts on Tumor Cell Proliferation and Tumor Prognosis in the Context of PI3K/PTEN Pathway Status.Cancers (Basel). 2021 Jul 28;13(15):3803. doi: 10.3390/cancers13153803. Cancers (Basel). 2021. PMID: 34359707 Free PMC article.
-
Role of Muscle-Specific Histone Methyltransferase (Smyd1) in Exercise-Induced Cardioprotection against Pathological Remodeling after Myocardial Infarction.Int J Mol Sci. 2020 Sep 23;21(19):7010. doi: 10.3390/ijms21197010. Int J Mol Sci. 2020. PMID: 32977624 Free PMC article.
-
Mitochondrial-derived peptides in cardiovascular disease: Novel insights and therapeutic opportunities.J Adv Res. 2024 Oct;64:99-115. doi: 10.1016/j.jare.2023.11.018. Epub 2023 Nov 24. J Adv Res. 2024. PMID: 38008175 Free PMC article. Review.
-
Lethal effects of mitochondria via microfluidics.Bioeng Transl Med. 2022 Dec 5;8(3):e10461. doi: 10.1002/btm2.10461. eCollection 2023 May. Bioeng Transl Med. 2022. PMID: 37206227 Free PMC article.
References
-
- Hafstad A.D., Nabeebaccus A.A., Shah A.M. Novel aspects of ROS signalling in heart failure. Basic Res Cardiol. 2013;108:359. - PubMed
-
- Rizzi E. β1-Adrenergic blockers exert antioxidant effects, reduce matrix metalloproteinase activity, and improve renovascular hypertension-induced cardiac hypertrophy. Free Radic Biol Med. 2014;73:308–317. - PubMed
-
- Kwon S.H., Pimentel D.R., Remondino A., Sawyer D.B., Colucci W.S. H(2)O(2) regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol. 2003;35:615–621. - PubMed
-
- Wang H.X. NADPH oxidases mediate a cellular "memory" of angiotensin II stress in hypertensive cardiac hypertrophy. Free Radic Biol Med. 2013;65:897–907. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
