

It's All about Timing: The Involvement of Kir4.1 Channel Regulation in Acute Ischemic Stroke Pathology
- PMID: 29503609
- PMCID: PMC5820340
- DOI: 10.3389/fncel.2018.00036
It's All about Timing: The Involvement of Kir4.1 Channel Regulation in Acute Ischemic Stroke Pathology
Abstract
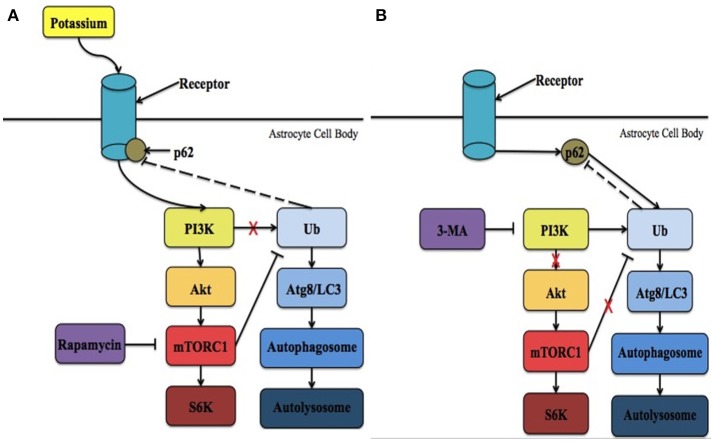
An acute ischemic stroke is characterized by the presence of a blood clot that limits blood flow to the brain resulting in subsequent neuronal loss. Acute stroke threatens neuronal survival, which relies heavily upon proper function of astrocytes. Neurons are more susceptible to cell death when an astrocyte is unable to carry out its normal functions in supporting the neuron in the area affected by the stroke (Rossi et al., 2007; Takano et al., 2009). For example, under normal conditions, astrocytes initially swell in response to changes in extracellular osmotic pressure and then reduce their regulatory volume in response to volume-activated potassium (K+) and chloride channels (Vella et al., 2015). This astroglial swelling may be overwhelmed, under ischemic conditions, due to the increased levels of glutamate and extracellular K+ (Lai et al., 2014; Vella et al., 2015). The increase in extracellular K+ contributes to neuronal damage and loss through the initiation of harmful secondary cascades (Nwaobi et al., 2016). Reducing the amount of extracellular K+ could, in theory, limit or prevent neuronal damage and loss resulting in an improved prognosis for individuals following ischemic stroke. Kir4.1, an inwardly rectifying K+ channel, has demonstrated an ability to regulate the rapid reuptake of this ion to return the cell to basal levels allowing it to fire again in rapid transmission (Sibille et al., 2015). Despite growing interest in this area, the underlying mechanism suggesting that neuroprotection could occur through modification of the Kir4.1 channel's activity has yet to be described. The purpose of this review is to examine the current literature and propose potential underlying mechanisms involving Kir4.1, specially the mammalian target of rapamycin (mTOR) and/or autophagic pathways, in the pathogenesis of ischemic stroke. The hope is that this review will instigate further investigation of Kir4.1 as a modulator of stroke pathology.
Keywords: Kir4.1; astrocytes; autophagy; ischemia; mammalian target of rapamycin.
Figures
Similar articles
-
Inwardly Rectifying Potassium Channel Kir4.1 as a Novel Modulator of BDNF Expression in Astrocytes.Int J Mol Sci. 2018 Oct 24;19(11):3313. doi: 10.3390/ijms19113313. Int J Mol Sci. 2018. PMID: 30356026 Free PMC article. Review.
-
Hyperglycemia reduces functional expression of astrocytic Kir4.1 channels and glial glutamate uptake.Neuroscience. 2015 Dec 3;310:216-23. doi: 10.1016/j.neuroscience.2015.09.044. Epub 2015 Sep 25. Neuroscience. 2015. PMID: 26404875 Free PMC article.
-
Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation.J Neurophysiol. 2006 Mar;95(3):1843-52. doi: 10.1152/jn.00996.2005. Epub 2005 Nov 23. J Neurophysiol. 2006. PMID: 16306174
-
Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes.Glia. 2007 Feb;55(3):274-81. doi: 10.1002/glia.20455. Glia. 2007. PMID: 17091490
-
Emerging Roles of Astrocyte Kir4.1 Channels in the Pathogenesis and Treatment of Brain Diseases.Int J Mol Sci. 2021 Sep 23;22(19):10236. doi: 10.3390/ijms221910236. Int J Mol Sci. 2021. PMID: 34638578 Free PMC article. Review.
Cited by
-
Dysregulation of Astrocyte Ion Homeostasis and Its Relevance for Stroke-Induced Brain Damage.Int J Mol Sci. 2021 May 26;22(11):5679. doi: 10.3390/ijms22115679. Int J Mol Sci. 2021. PMID: 34073593 Free PMC article. Review.
-
New Candidates for Biomarkers and Drug Targets of Ischemic Stroke-A First Dynamic LC-MS Human Serum Proteomic Study.J Clin Med. 2022 Jan 11;11(2):339. doi: 10.3390/jcm11020339. J Clin Med. 2022. PMID: 35054033 Free PMC article.
-
VU6036720: The First Potent and Selective In Vitro Inhibitor of Heteromeric Kir4.1/5.1 Inward Rectifier Potassium Channels.Mol Pharmacol. 2022 May;101(5):357-370. doi: 10.1124/molpharm.121.000464. Epub 2022 Mar 3. Mol Pharmacol. 2022. PMID: 35246480 Free PMC article.
-
MK-801 attenuates lesion expansion following acute brain injury in rats: a meta-analysis.Neural Regen Res. 2019 Nov;14(11):1919-1931. doi: 10.4103/1673-5374.259619. Neural Regen Res. 2019. PMID: 31290450 Free PMC article.
-
A Purinergic P2 Receptor Family-Mediated Increase in Thrombospondin-1 Bolsters Synaptic Density and Epileptic Seizure Activity in the Amygdala-Kindling Rat Model.Front Cell Neurosci. 2018 Oct 1;12:302. doi: 10.3389/fncel.2018.00302. eCollection 2018. Front Cell Neurosci. 2018. PMID: 30386206 Free PMC article.
References
-
- Bond C. T., Pessia M., Xia X. M., Lagrutta A., Kavanaugh M. P., Adelman J. P. (1994). Cloning and expression of a family of inward rectifier potassium channels. Recept. Channels 2, 183–191. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous