

Single-cell developmental classification of B cell precursor acute lymphoblastic leukemia at diagnosis reveals predictors of relapse
- PMID: 29505032
- PMCID: PMC5953207
- DOI: 10.1038/nm.4505
Single-cell developmental classification of B cell precursor acute lymphoblastic leukemia at diagnosis reveals predictors of relapse
Abstract
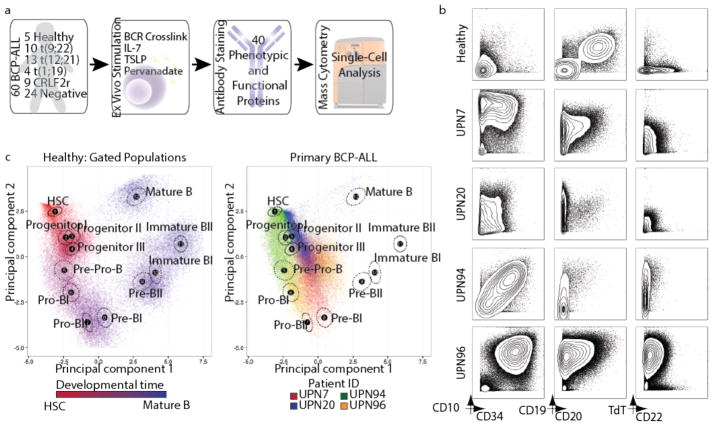
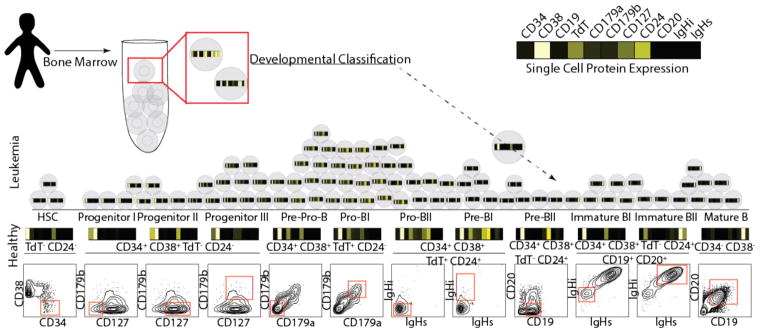
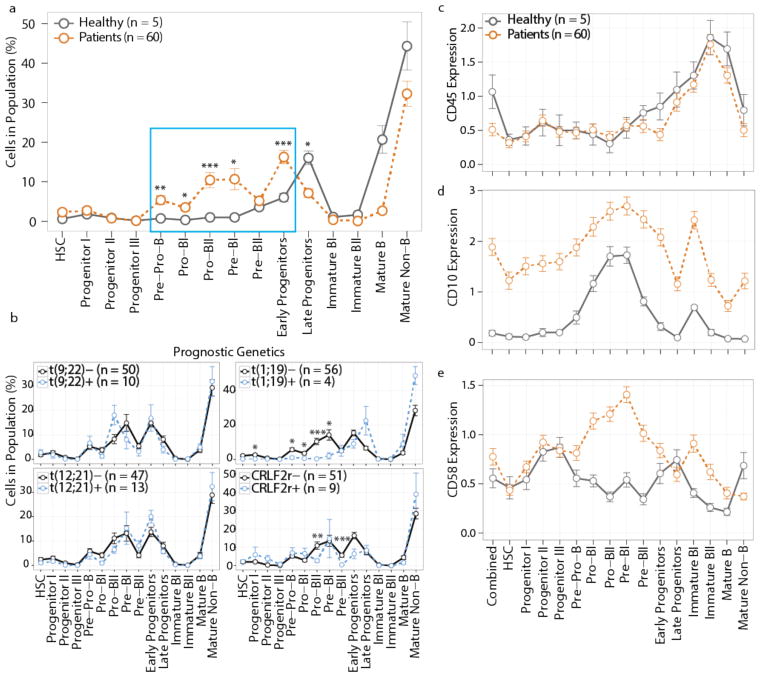
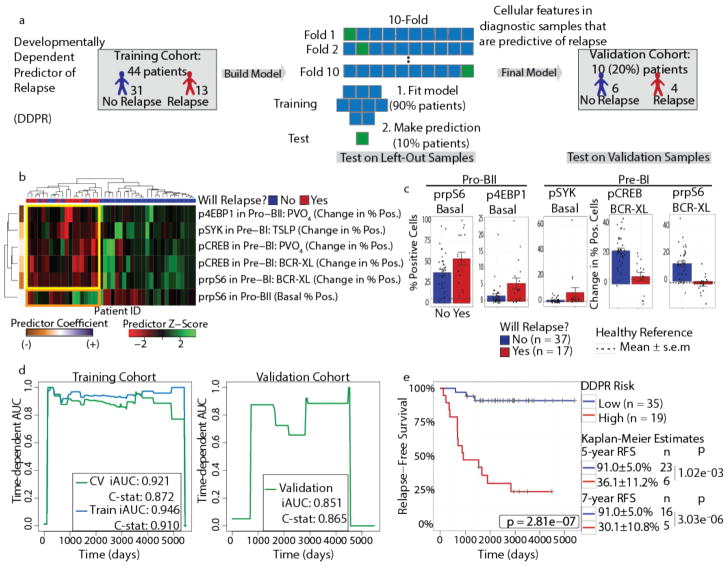
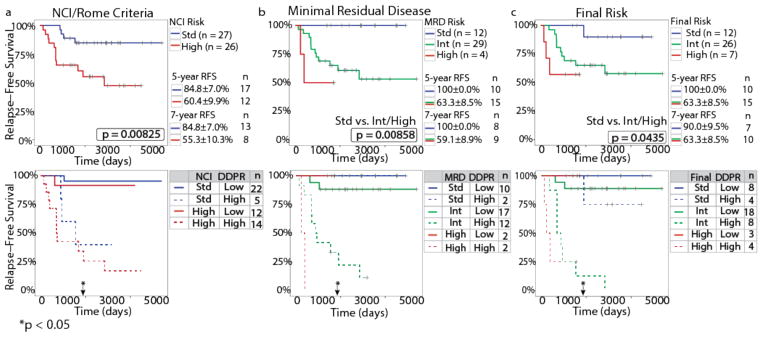
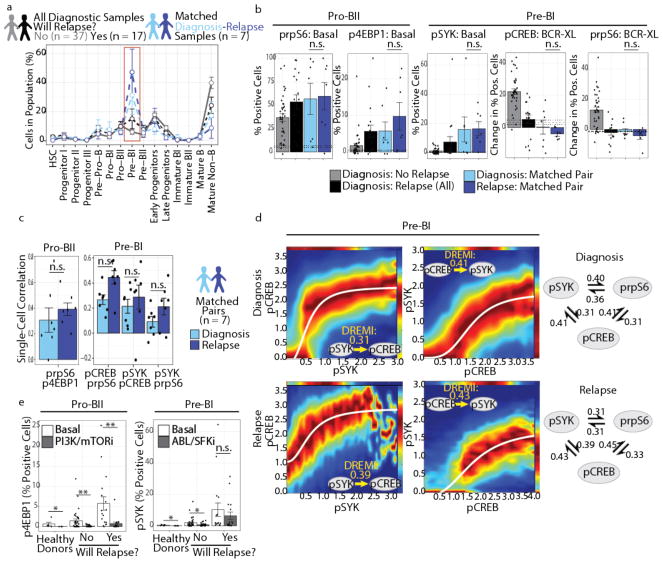
Insight into the cancer cell populations that are responsible for relapsed disease is needed to improve outcomes. Here we report a single-cell-based study of B cell precursor acute lymphoblastic leukemia at diagnosis that reveals hidden developmentally dependent cell signaling states that are uniquely associated with relapse. By using mass cytometry we simultaneously quantified 35 proteins involved in B cell development in 60 primary diagnostic samples. Each leukemia cell was then matched to its nearest healthy B cell population by a developmental classifier that operated at the single-cell level. Machine learning identified six features of expanded leukemic populations that were sufficient to predict patient relapse at diagnosis. These features implicated the pro-BII subpopulation of B cells with activated mTOR signaling, and the pre-BI subpopulation of B cells with activated and unresponsive pre-B cell receptor signaling, to be associated with relapse. This model, termed 'developmentally dependent predictor of relapse' (DDPR), significantly improves currently established risk stratification methods. DDPR features exist at diagnosis and persist at relapse. By leveraging a data-driven approach, we demonstrate the predictive value of single-cell 'omics' for patient stratification in a translational setting and provide a framework for its application to human cancer.
Conflict of interest statement
S.C.B. and G.P.N. are paid consultants for Fluidigm, the manufacturer that produced some of the reagents and instrumentation used in this manuscript.
Figures
Comment in
-
Leukaemia: Powers of prediction.Nat Rev Cancer. 2018 May;18(5):372-373. doi: 10.1038/nrc.2018.28. Epub 2018 Apr 3. Nat Rev Cancer. 2018. PMID: 29610489 No abstract available.
-
Predicting leukemia relapse.Nat Med. 2018 Apr 10;24(4):385-387. doi: 10.1038/nm.4529. Nat Med. 2018. PMID: 29634681 No abstract available.
References
-
- Anderson K, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2010;469:356–361. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
