

Hereditary cancer genes are highly susceptible to splicing mutations
- PMID: 29505604
- PMCID: PMC5854443
- DOI: 10.1371/journal.pgen.1007231
Hereditary cancer genes are highly susceptible to splicing mutations
Abstract
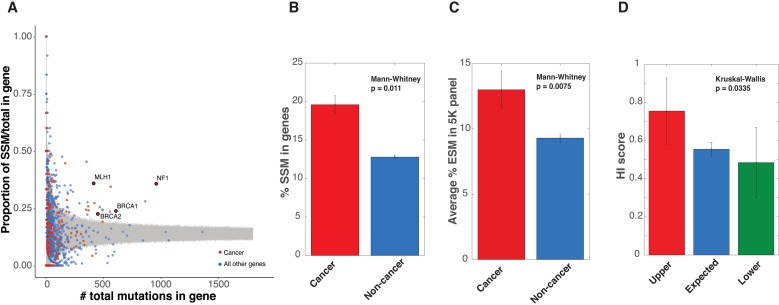
Substitutions that disrupt pre-mRNA splicing are a common cause of genetic disease. On average, 13.4% of all hereditary disease alleles are classified as splicing mutations mapping to the canonical 5' and 3' splice sites. However, splicing mutations present in exons and deeper intronic positions are vastly underreported. A recent re-analysis of coding mutations in exon 10 of the Lynch Syndrome gene, MLH1, revealed an extremely high rate (77%) of mutations that lead to defective splicing. This finding is confirmed by extending the sampling to five other exons in the MLH1 gene. Further analysis suggests a more general phenomenon of defective splicing driving Lynch Syndrome. Of the 36 mutations tested, 11 disrupted splicing. Furthermore, analyzing past reports suggest that MLH1 mutations in canonical splice sites also occupy a much higher fraction (36%) of total mutations than expected. When performing a comprehensive analysis of splicing mutations in human disease genes, we found that three main causal genes of Lynch Syndrome, MLH1, MSH2, and PMS2, belonged to a class of 86 disease genes which are enriched for splicing mutations. Other cancer genes were also enriched in the 86 susceptible genes. The enrichment of splicing mutations in hereditary cancers strongly argues for additional priority in interpreting clinical sequencing data in relation to cancer and splicing.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures




References
-
- Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393 - DOI - PMC - PubMed
-
- Fu W, O'Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493(7431):216–20. doi: 10.1038/nature11690 - DOI - PMC - PubMed
-
- Lim KH, Fairbrother WG. Spliceman—a computational web server that predicts sequence variations in pre-mRNA splicing. Bioinformatics. 2012;28(7):1031–2. doi: 10.1093/bioinformatics/bts074 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
