

Murine knockin model for progranulin-deficient frontotemporal dementia with nonsense-mediated mRNA decay
- PMID: 29511098
- PMCID: PMC5866607
- DOI: 10.1073/pnas.1722344115
Murine knockin model for progranulin-deficient frontotemporal dementia with nonsense-mediated mRNA decay
Abstract
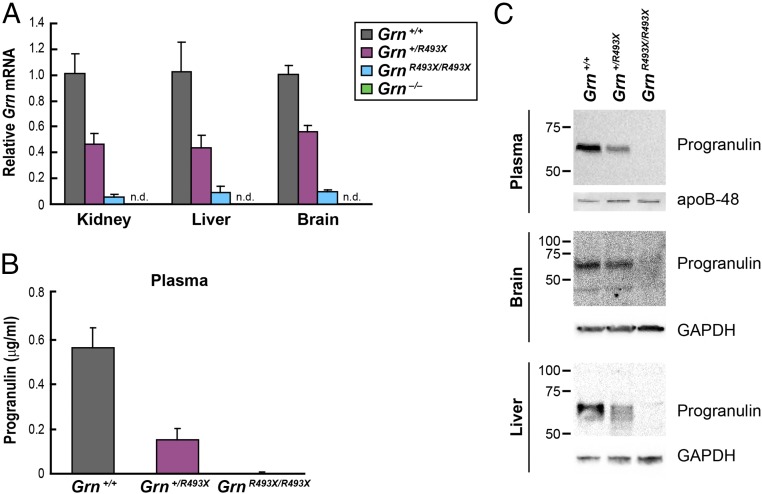
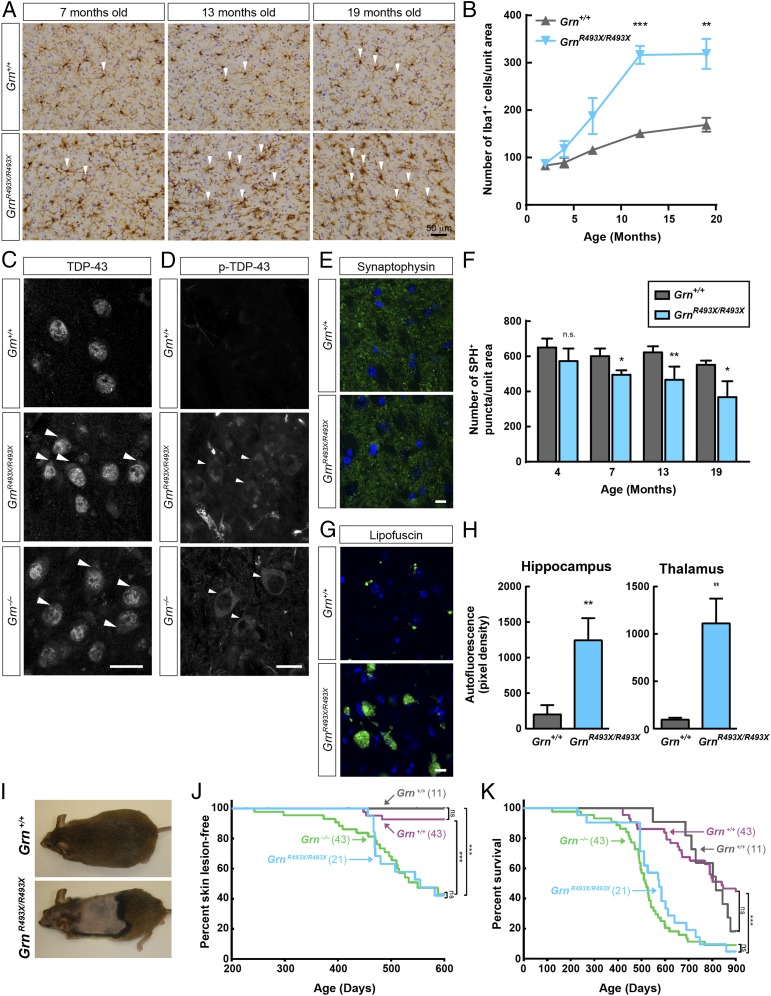
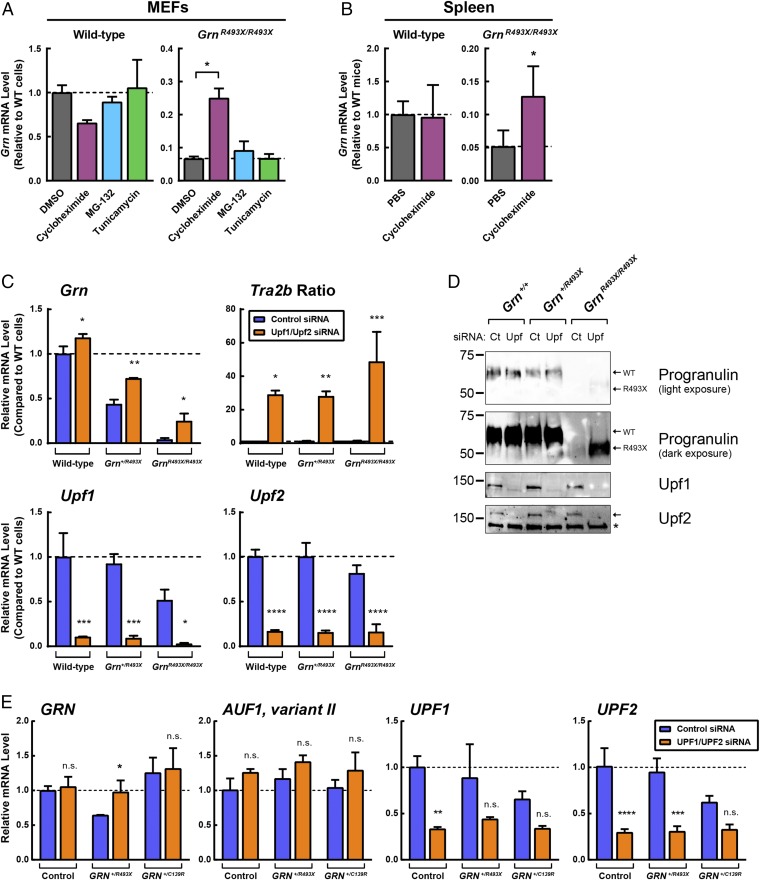
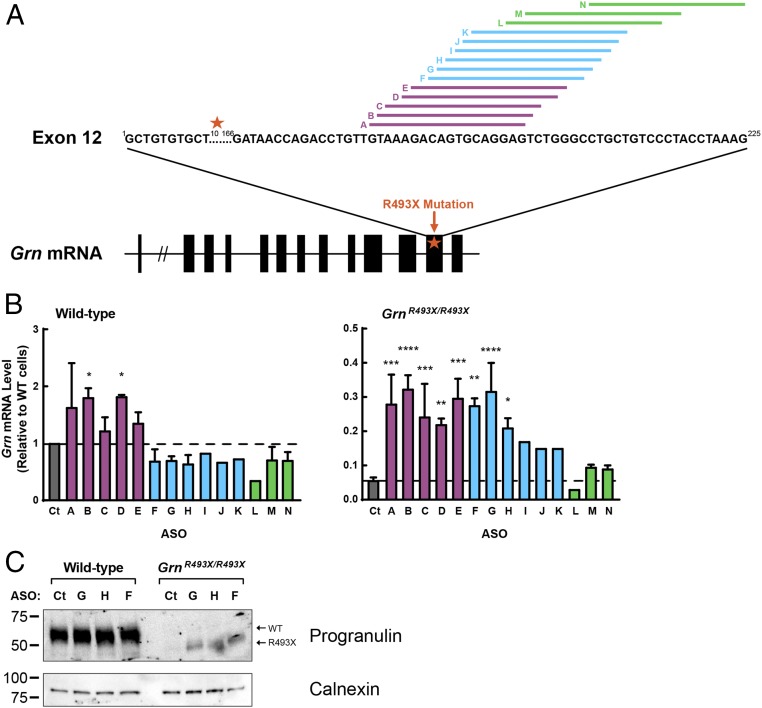
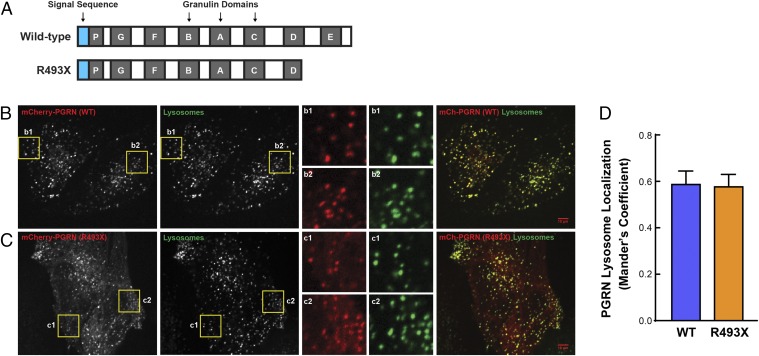
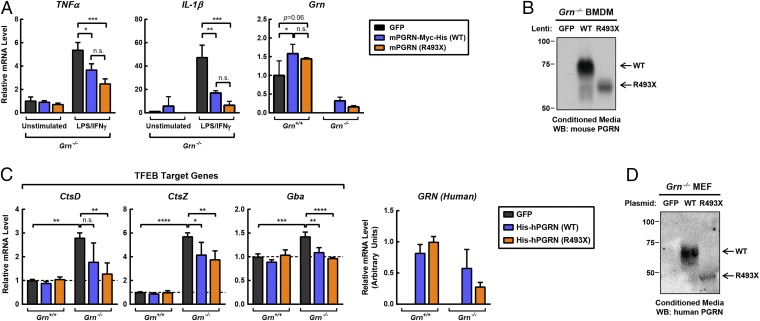
Frontotemporal dementia (FTD) is the most common neurodegenerative disorder in individuals under age 60 and has no treatment or cure. Because many cases of FTD result from GRN nonsense mutations, an animal model for this type of mutation is highly desirable for understanding pathogenesis and testing therapies. Here, we generated and characterized GrnR493X knockin mice, which model the most common human GRN mutation, a premature stop codon at arginine 493 (R493X). Homozygous GrnR493X mice have markedly reduced Grn mRNA levels, lack detectable progranulin protein, and phenocopy Grn knockout mice, with CNS microgliosis, cytoplasmic TDP-43 accumulation, reduced synaptic density, lipofuscinosis, hyperinflammatory macrophages, excessive grooming behavior, and reduced survival. Inhibition of nonsense-mediated mRNA decay (NMD) by genetic, pharmacological, or antisense oligonucleotide-based approaches showed that NMD contributes to the reduced mRNA levels in GrnR493X mice and cell lines and in fibroblasts from patients containing the GRNR493X mutation. Moreover, the expressed truncated R493X mutant protein was functional in several assays in progranulin-deficient cells. Together, these findings establish a murine model for in vivo testing of NMD inhibition or other therapies as potential approaches for treating progranulin deficiency caused by the R493X mutation.
Keywords: frontotemporal dementia; lysosome; neurodegeneration; nonsense-mediated mRNA decay; progranulin.
Copyright © 2018 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Baker M, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. - PubMed
-
- Cruts M, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. - PubMed
-
- Gass J, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. - PubMed
-
- Cruts M. 2012 The AD & FTLD mutation database. Available at www.molgen.vib-ua.be/FTDMutations. Accessed November 8, 2016.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
