

Phenotype of CNTNAP1: a study of patients demonstrating a specific severe congenital hypomyelinating neuropathy with survival beyond infancy
- PMID: 29511323
- PMCID: PMC5974240
- DOI: 10.1038/s41431-018-0110-x
Phenotype of CNTNAP1: a study of patients demonstrating a specific severe congenital hypomyelinating neuropathy with survival beyond infancy
Abstract
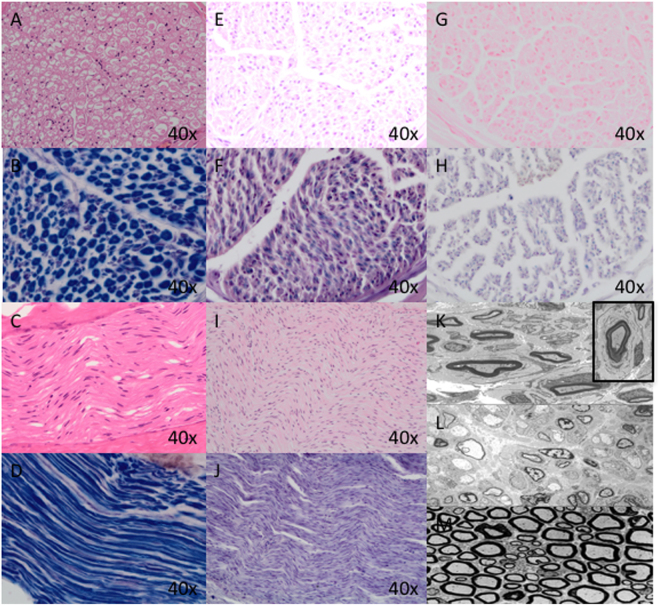
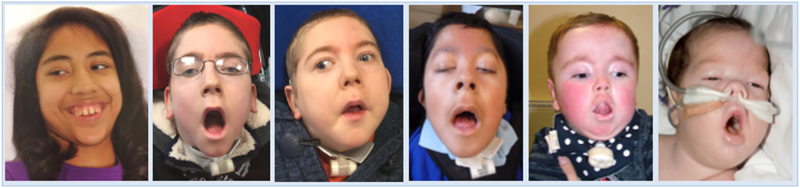
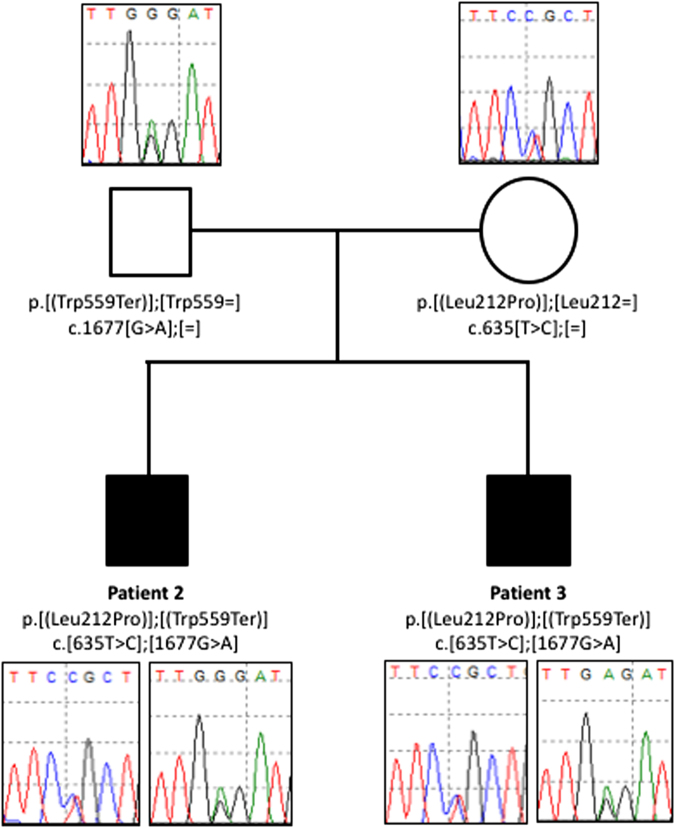
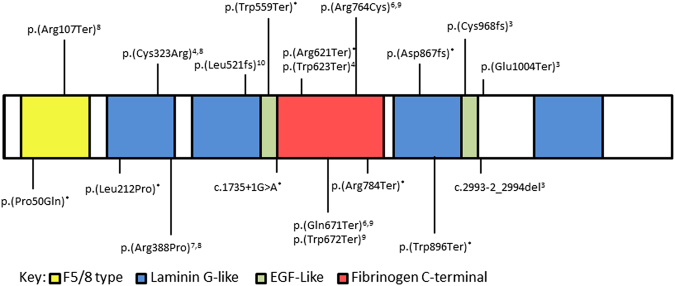
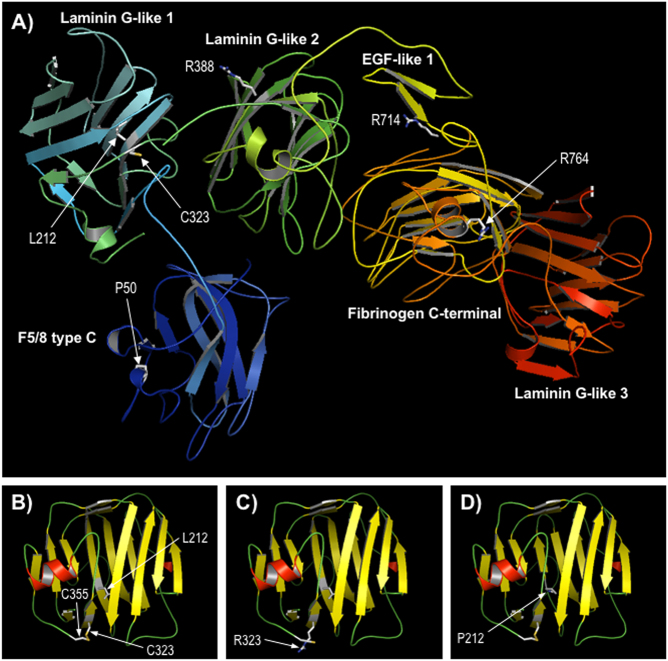
CHN is genetically heterogeneous and its genetic basis is difficult to determine on features alone. CNTNAP1 encodes CASPR, integral in the paranodal junction high molecular mass complex. Nineteen individuals with biallelic variants have been described in association with severe congenital hypomyelinating neuropathy, respiratory compromise, profound intellectual disability and death within the first year. We report 7 additional patients ascertained through exome sequencing. We identified 9 novel CNTNAP1 variants in 6 families: three missense variants, four nonsense variants, one frameshift variant and one splice site variant. Significant polyhydramnios occurred in 6/7 pregnancies. Severe respiratory compromise was seen in 6/7 (tracheostomy in 5). A complex neurological phenotype was seen in all patients who had marked brain hypomyelination/demyelination and profound developmental delay. Additional neurological findings included cranial nerve compromise: orobulbar dysfunction in 5/7, facial nerve weakness in 4/7 and vocal cord paresis in 5/7. Dystonia occurred in 2/7 patients and limb contractures in 5/7. All had severe gastroesophageal reflux, and a gastrostomy was required in 5/7. In contrast to most previous reports, only one patient died in the first year of life. Protein modelling was performed for all detected CNTNAP1 variants. We propose a genotype-phenotype correlation, whereby hypomorphic missense variants partially ameliorate the phenotype, prolonging survival. This study suggests that biallelic variants in CNTNAP1 cause a distinct recognisable syndrome, which is not caused by other genes associated with CHN. Neonates presenting with this phenotype will benefit from early genetic definition to inform clinical management and enable essential genetic counselling for their families.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
