

EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma
- PMID: 29513652
- PMCID: PMC6318124
- DOI: 10.1038/nature25748
EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma
Erratum in
-
Author Correction: EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma.Nature. 2018 Jul;559(7715):E11. doi: 10.1038/s41586-018-0230-z. Nature. 2018. PMID: 29950716
Abstract
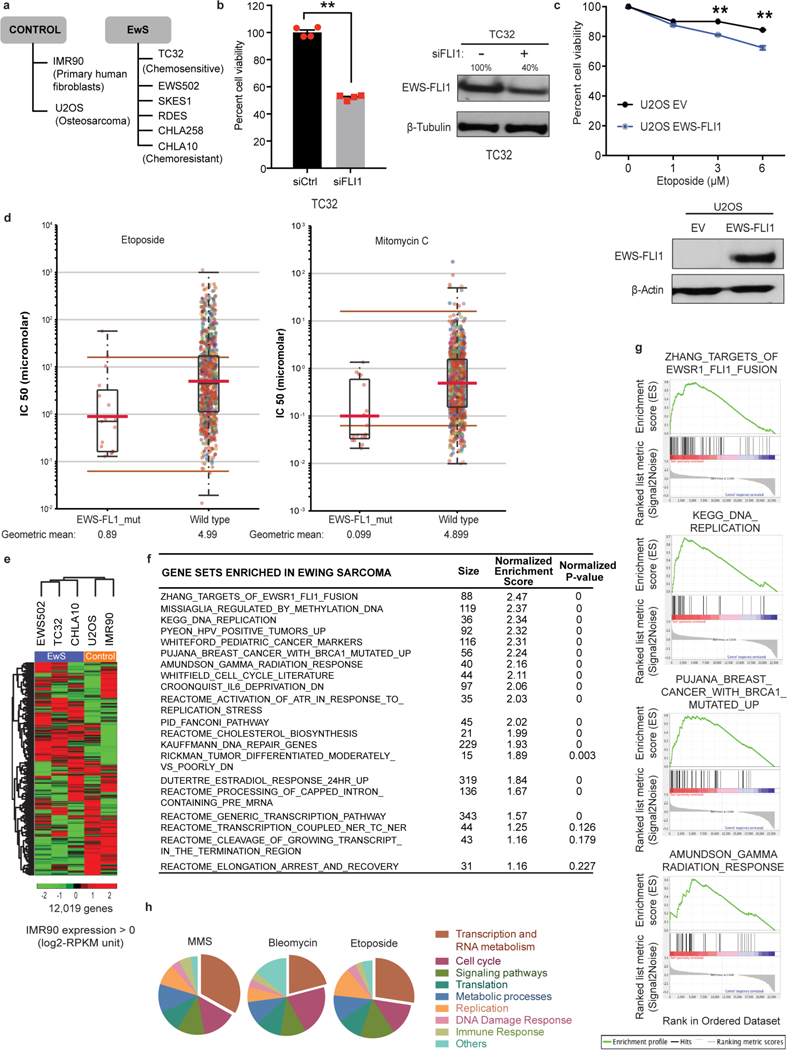
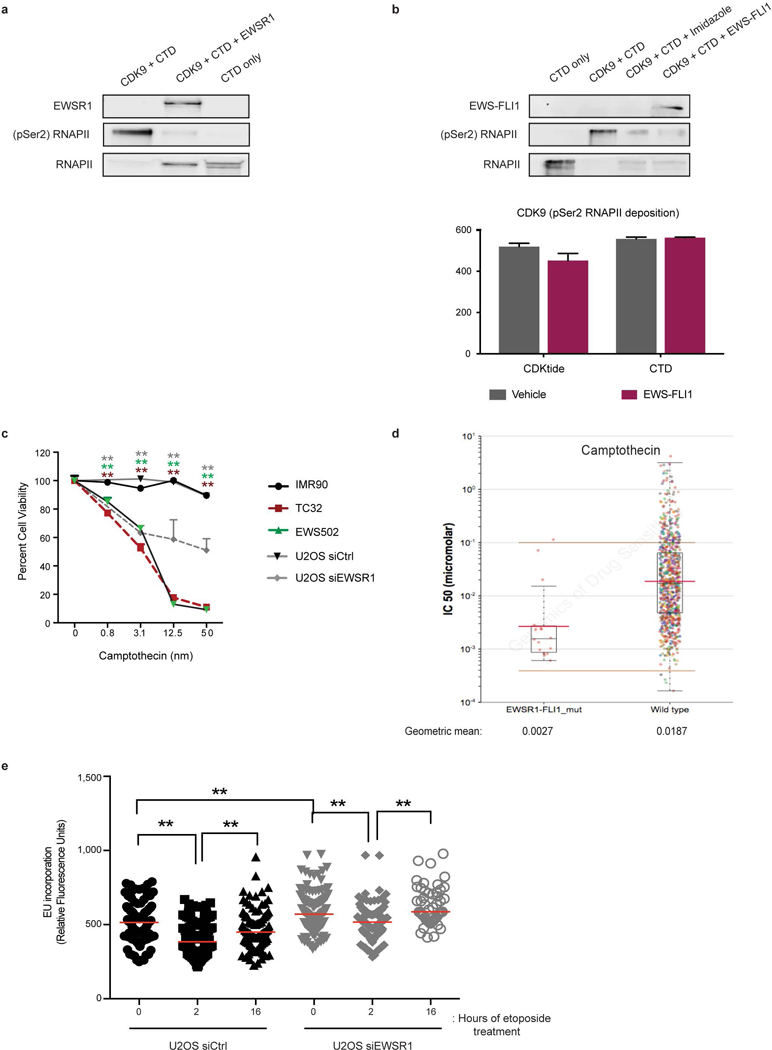
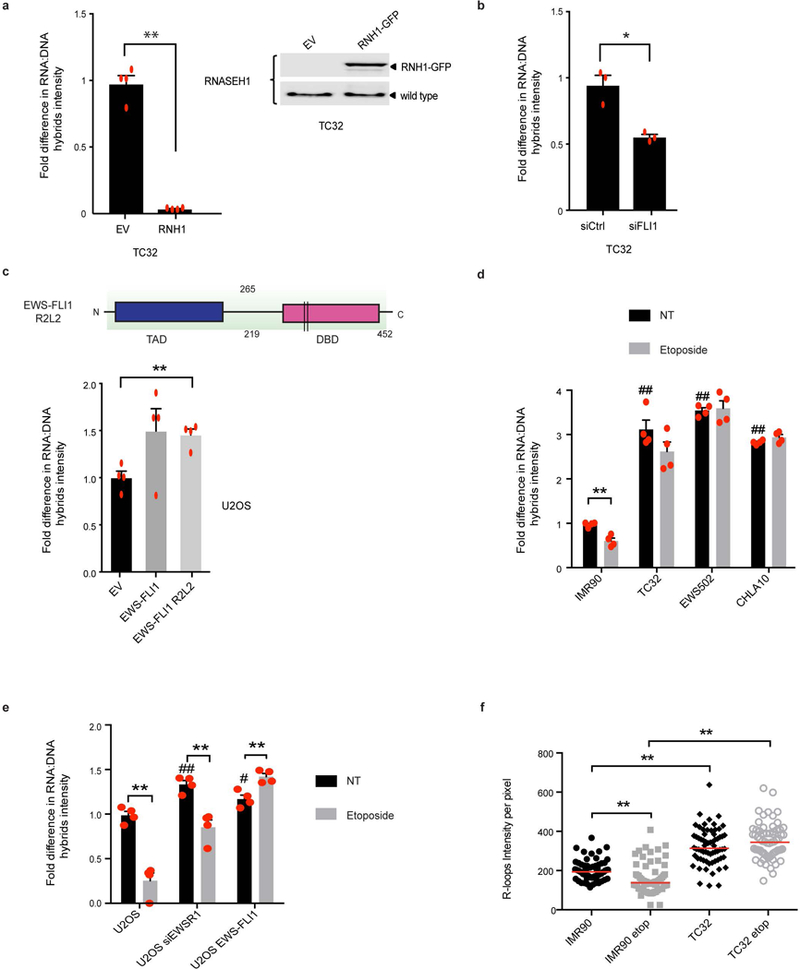
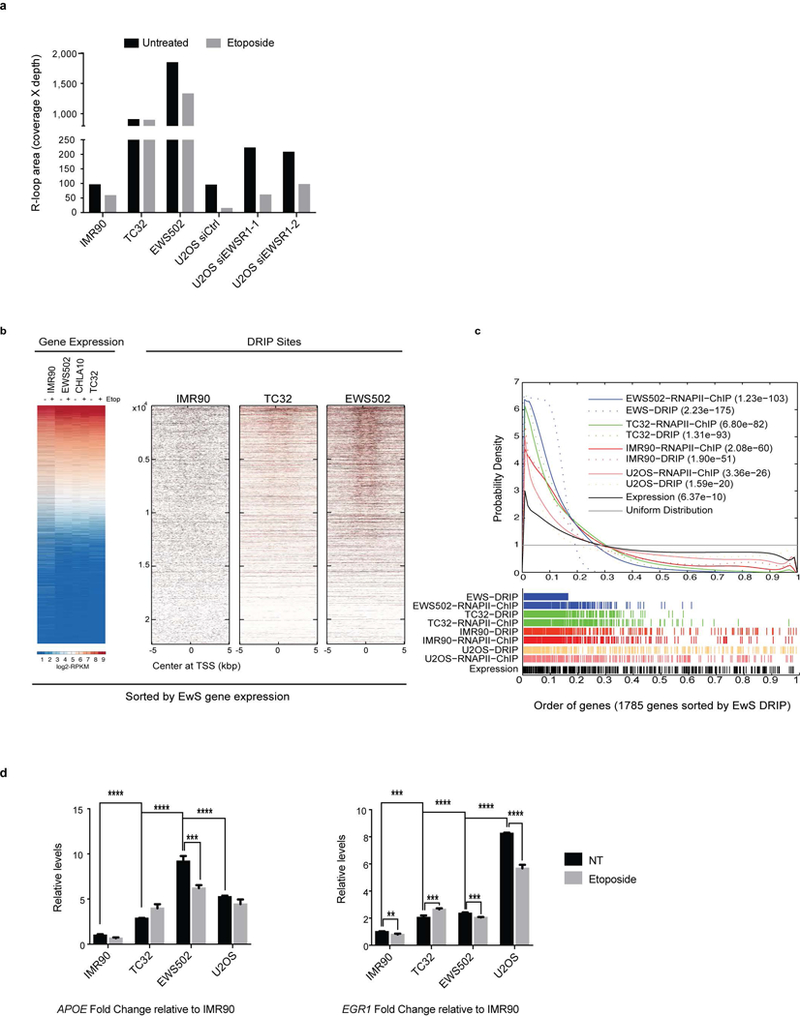
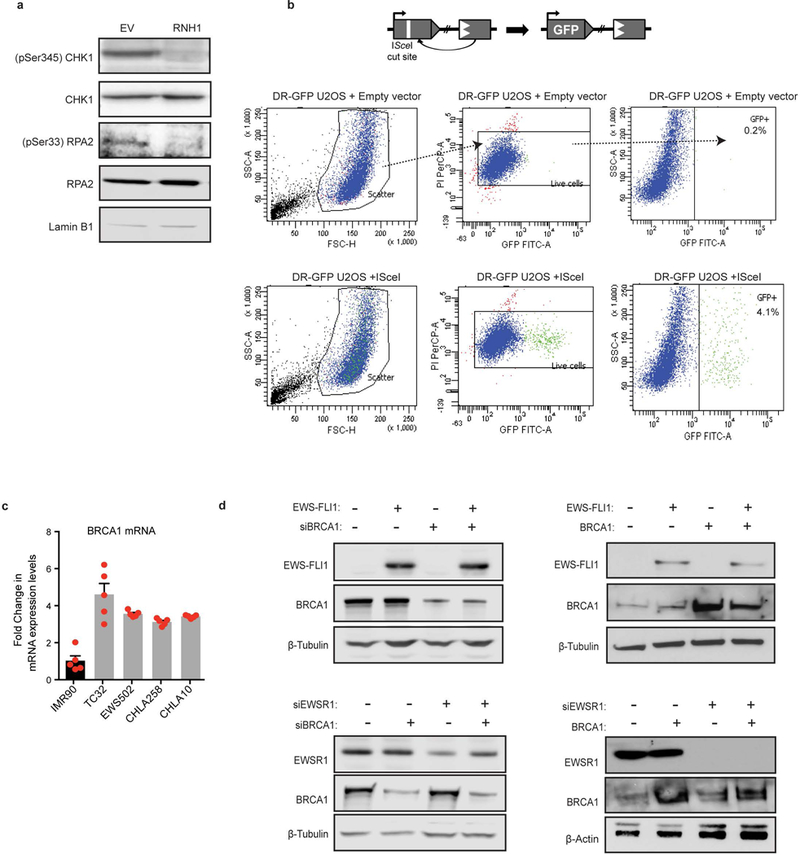
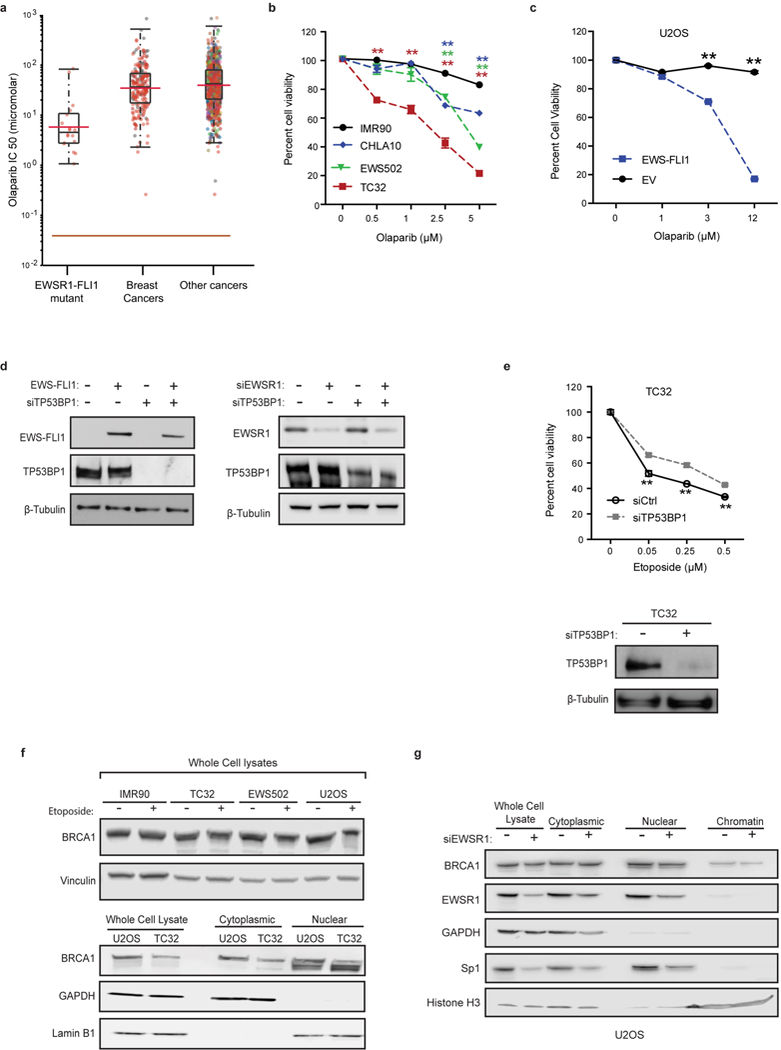
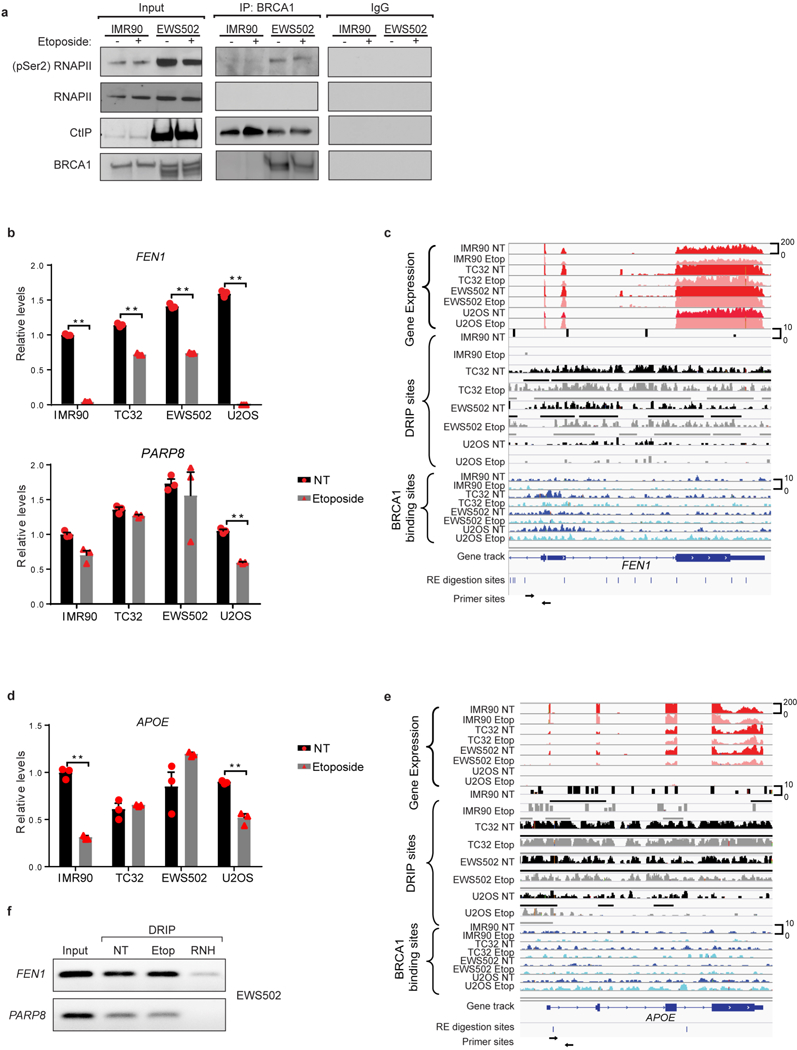
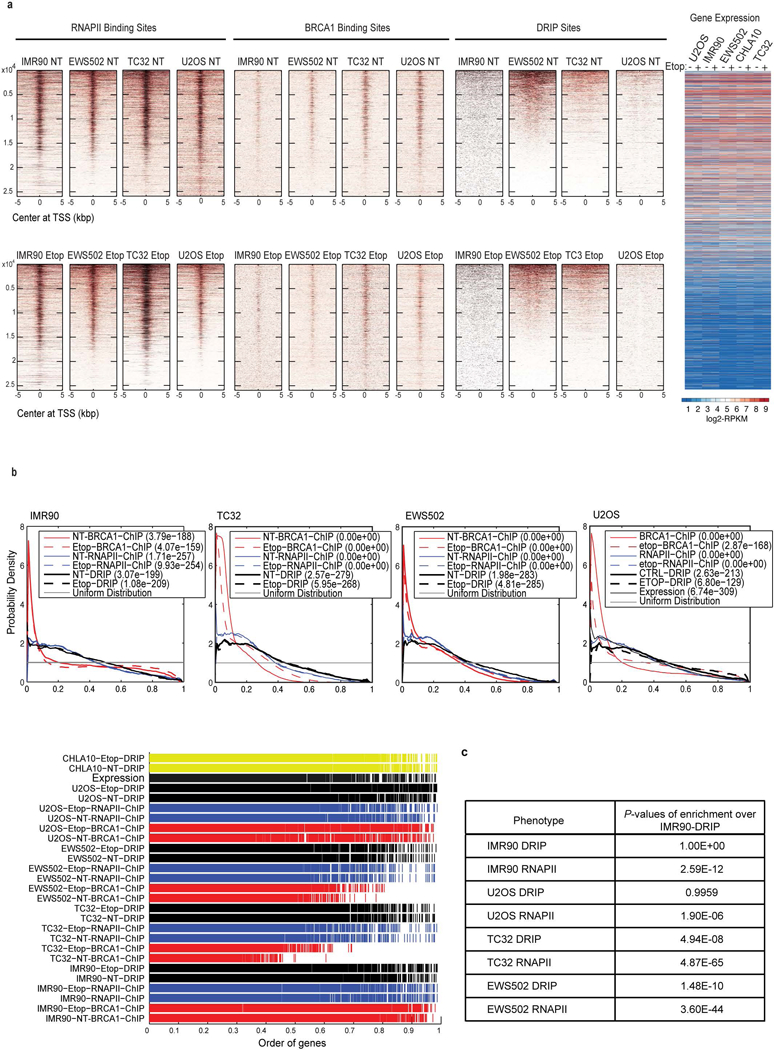
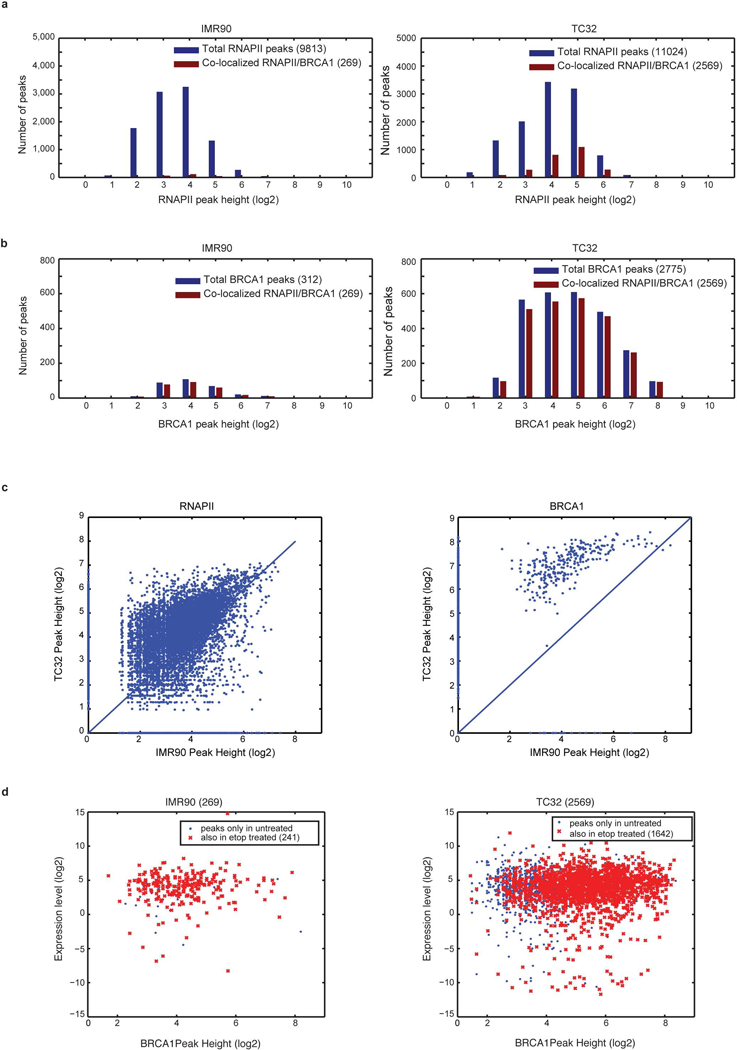
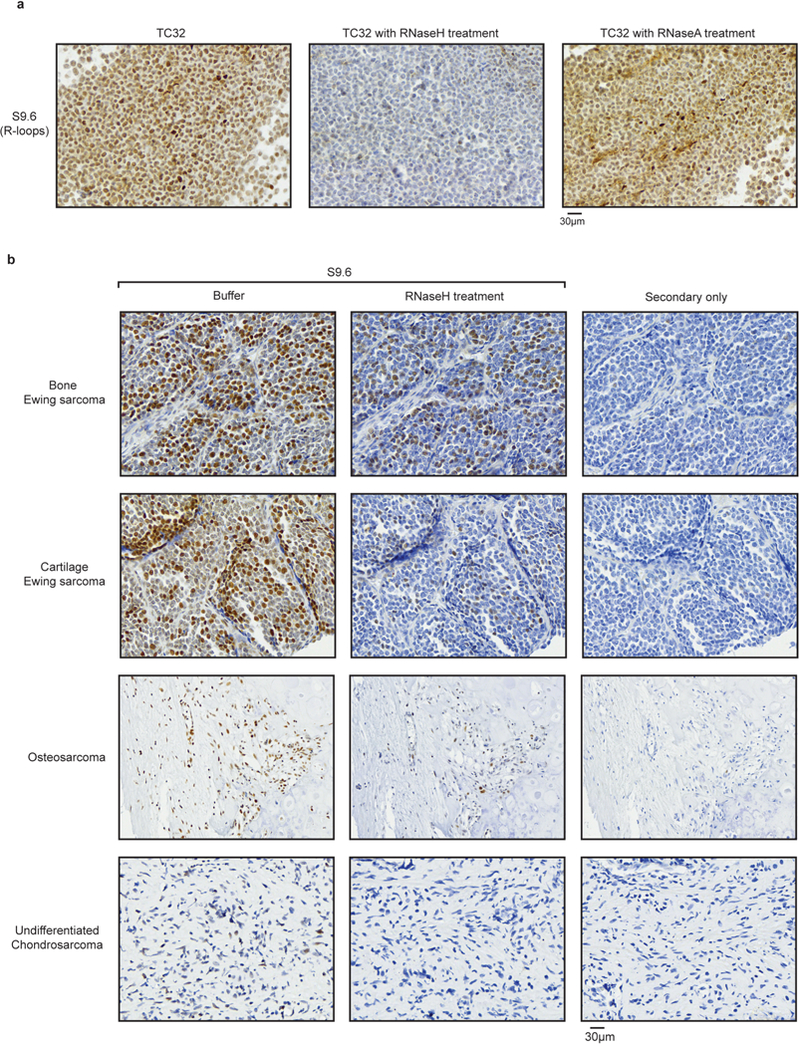
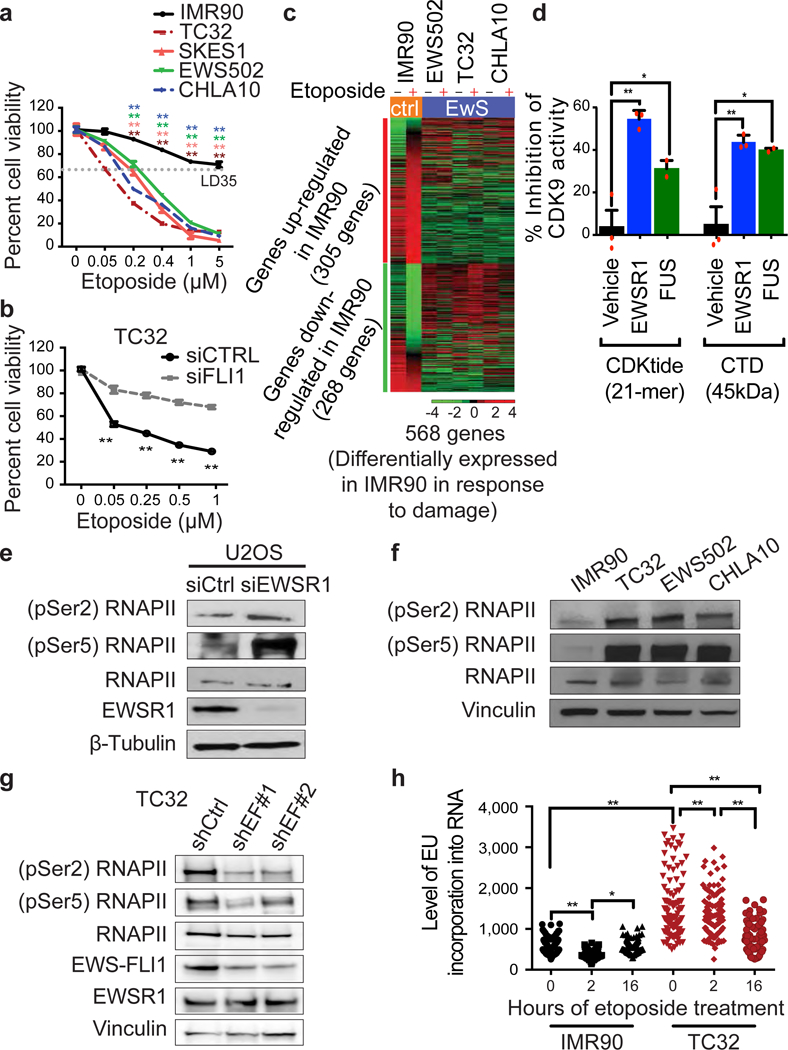
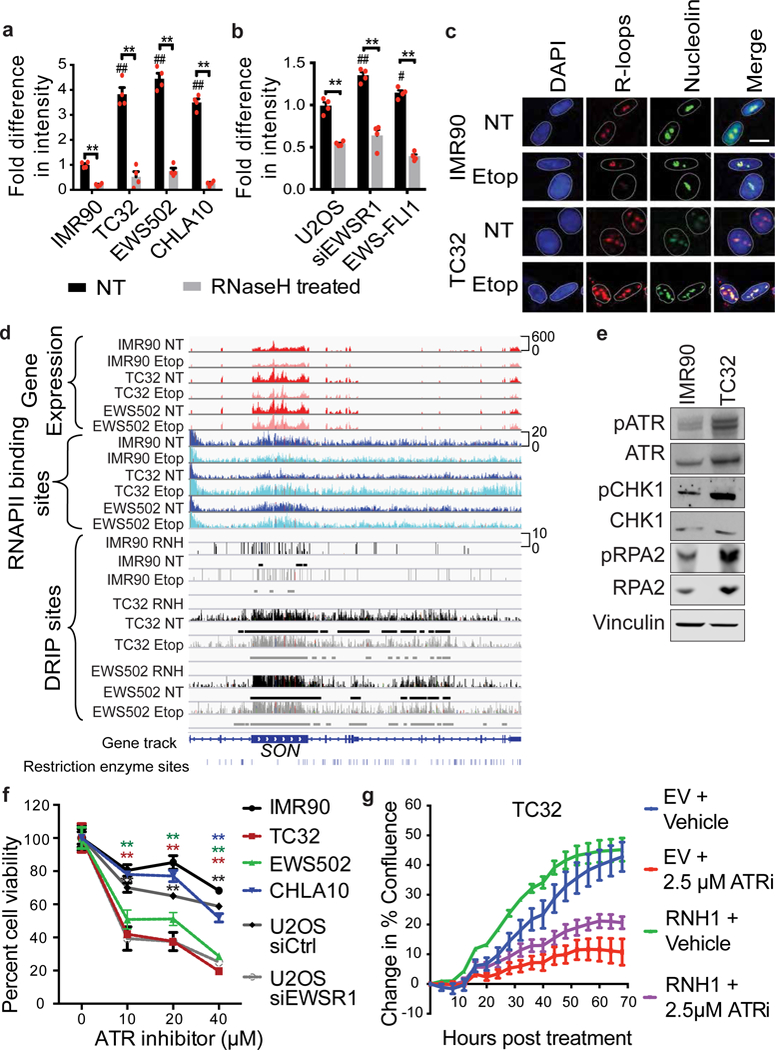
Ewing sarcoma is an aggressive paediatric cancer of the bone and soft tissue. It results from a chromosomal translocation, predominantly t(11;22)(q24:q12), that fuses the N-terminal transactivation domain of the constitutively expressed EWSR1 protein with the C-terminal DNA binding domain of the rarely expressed FLI1 protein. Ewing sarcoma is highly sensitive to genotoxic agents such as etoposide, but the underlying molecular basis of this sensitivity is unclear. Here we show that Ewing sarcoma cells display alterations in regulation of damage-induced transcription, accumulation of R-loops and increased replication stress. In addition, homologous recombination is impaired in Ewing sarcoma owing to an enriched interaction between BRCA1 and the elongating transcription machinery. Finally, we uncover a role for EWSR1 in the transcriptional response to damage, suppressing R-loops and promoting homologous recombination. Our findings improve the current understanding of EWSR1 function, elucidate the mechanistic basis of the sensitivity of Ewing sarcoma to chemotherapy (including PARP1 inhibitors) and highlight a class of BRCA-deficient-like tumours.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
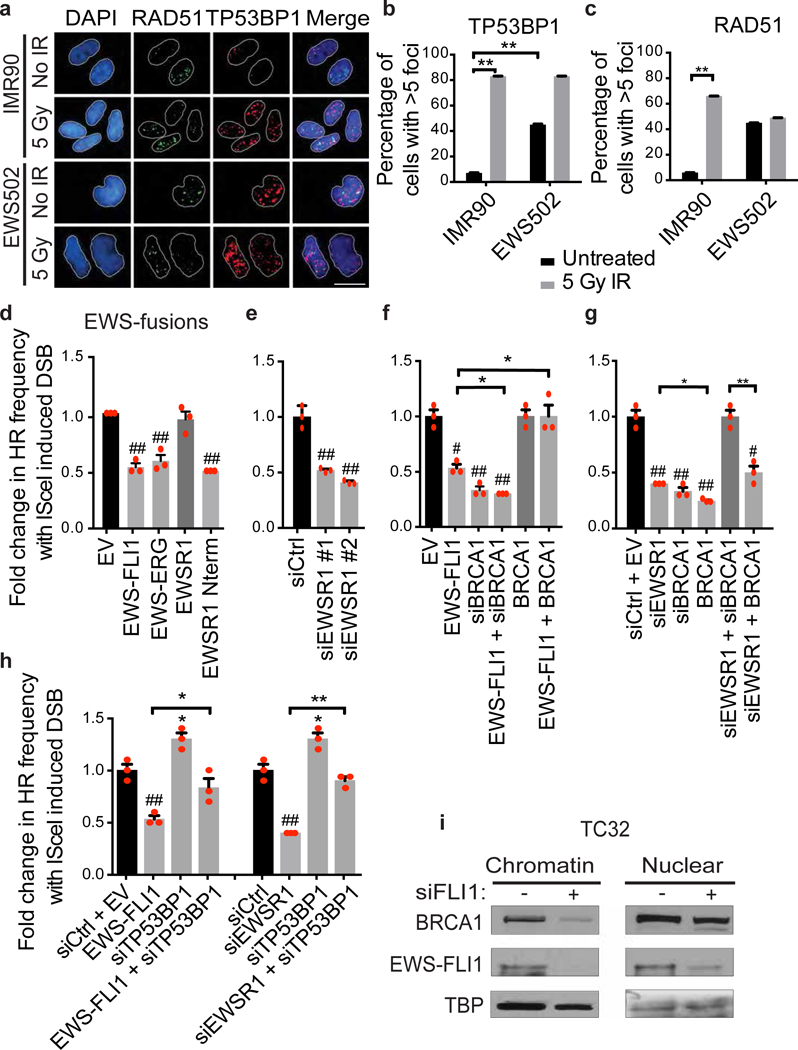
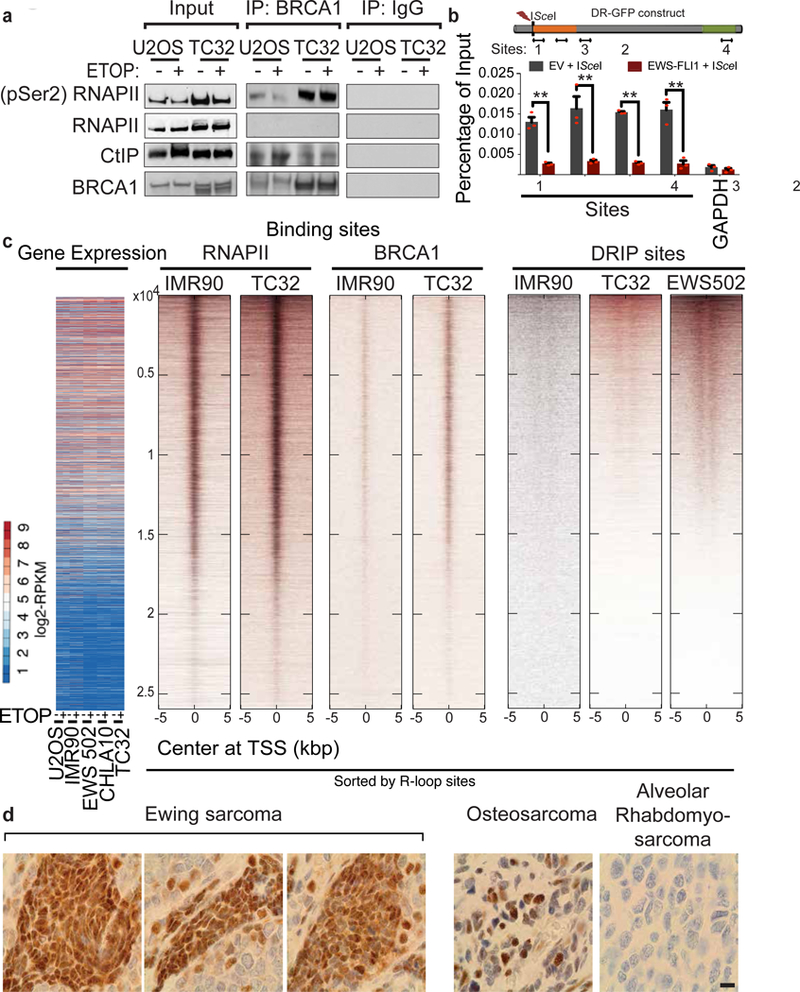
References
-
- Turc-Carel C, Philip I, Berger MP, Philip T & Lenoir GM Chromosome study of Ewing’s sarcoma (ES) cell lines. Consistency of a reciprocal translocation t(11;22)(q24;q12). Cancer Genet. Cytogenet. 12, 1–19 (1984). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
