

Developmental diversification of cortical inhibitory interneurons
- PMID: 29513653
- PMCID: PMC6052457
- DOI: 10.1038/nature25999
Developmental diversification of cortical inhibitory interneurons
Abstract
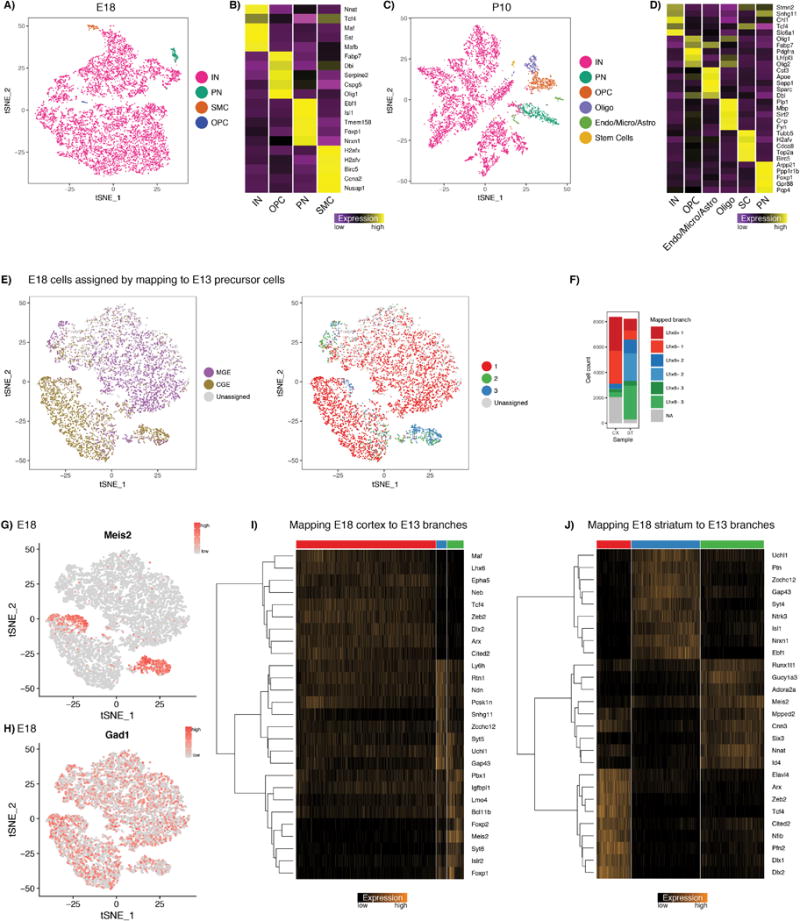
Diverse subsets of cortical interneurons have vital roles in higher-order brain functions. To investigate how this diversity is generated, here we used single-cell RNA sequencing to profile the transcriptomes of mouse cells collected along a developmental time course. Heterogeneity within mitotic progenitors in the ganglionic eminences is driven by a highly conserved maturation trajectory, alongside eminence-specific transcription factor expression that seeds the emergence of later diversity. Upon becoming postmitotic, progenitors diverge and differentiate into transcriptionally distinct states, including an interneuron precursor state. By integrating datasets across developmental time points, we identified shared sources of transcriptomic heterogeneity between adult interneurons and their precursors, and uncovered the embryonic emergence of cardinal interneuron subtypes. Our analysis revealed that the transcription factor Mef2c, which is linked to various neuropsychiatric and neurodevelopmental disorders, delineates early precursors of parvalbumin-expressing neurons, and is essential for their development. These findings shed new light on the molecular diversification of early inhibitory precursors, and identify gene modules that may influence the specification of human interneuron subtypes.
Figures













Comment in
-
A mixed model of neuronal diversity.Nature. 2018 Mar 22;555(7697):452-454. doi: 10.1038/d41586-018-02539-4. Nature. 2018. PMID: 29565398 No abstract available.
References
-
- Yun K, Garel S, Fischman S, Rubenstein JLR. Patterning of the lateral ganglionic eminence by the Gsh1 and Gsh2 homeobox genes regulates striatal and olfactory bulb histogenesis and the growth of axons through the basal ganglia. J Comp Neurol. 2003;461:151–165. - PubMed
-
- Shimamura K, Hartigan DJ, Martinez S, Puelles L, Rubenstein JL. Longitudinal organization of the anterior neural plate and neural tube. Development. 1995;121:3923–3933. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
