

Loss of CLN7 results in depletion of soluble lysosomal proteins and impaired mTOR reactivation
- PMID: 29514215
- PMCID: PMC5932567
- DOI: 10.1093/hmg/ddy076
Loss of CLN7 results in depletion of soluble lysosomal proteins and impaired mTOR reactivation
Abstract
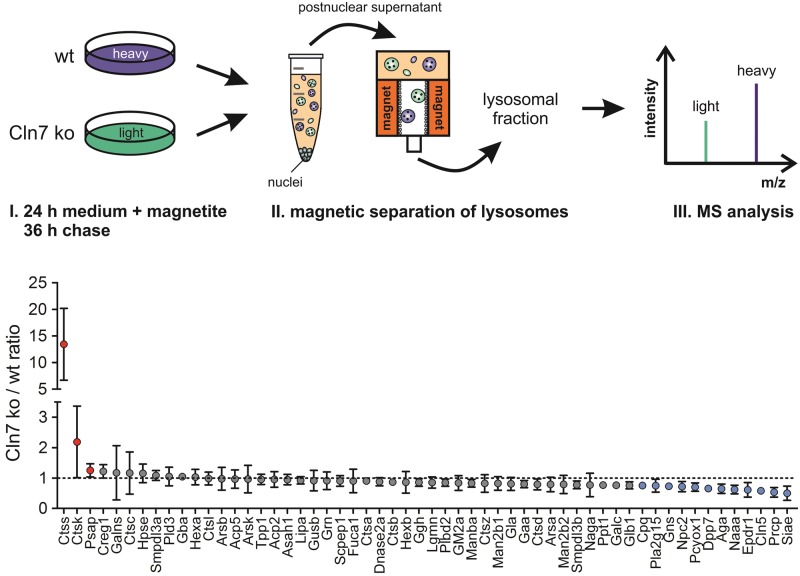
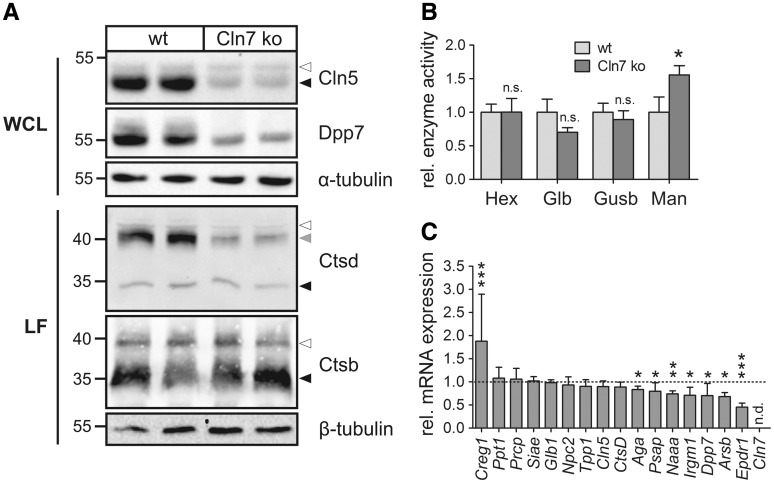
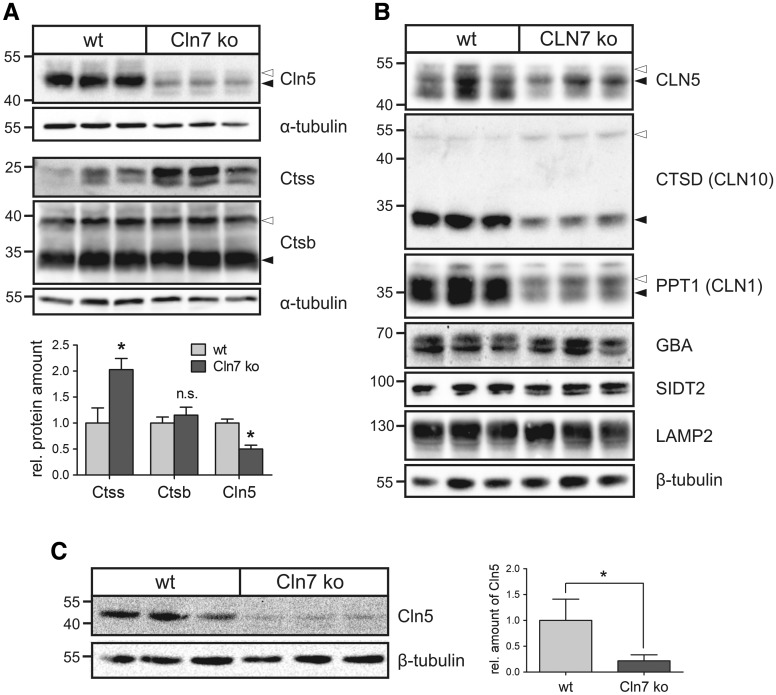
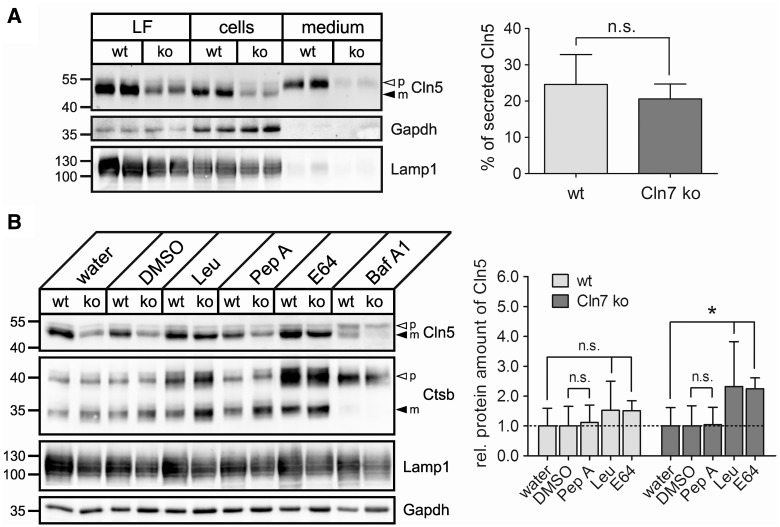
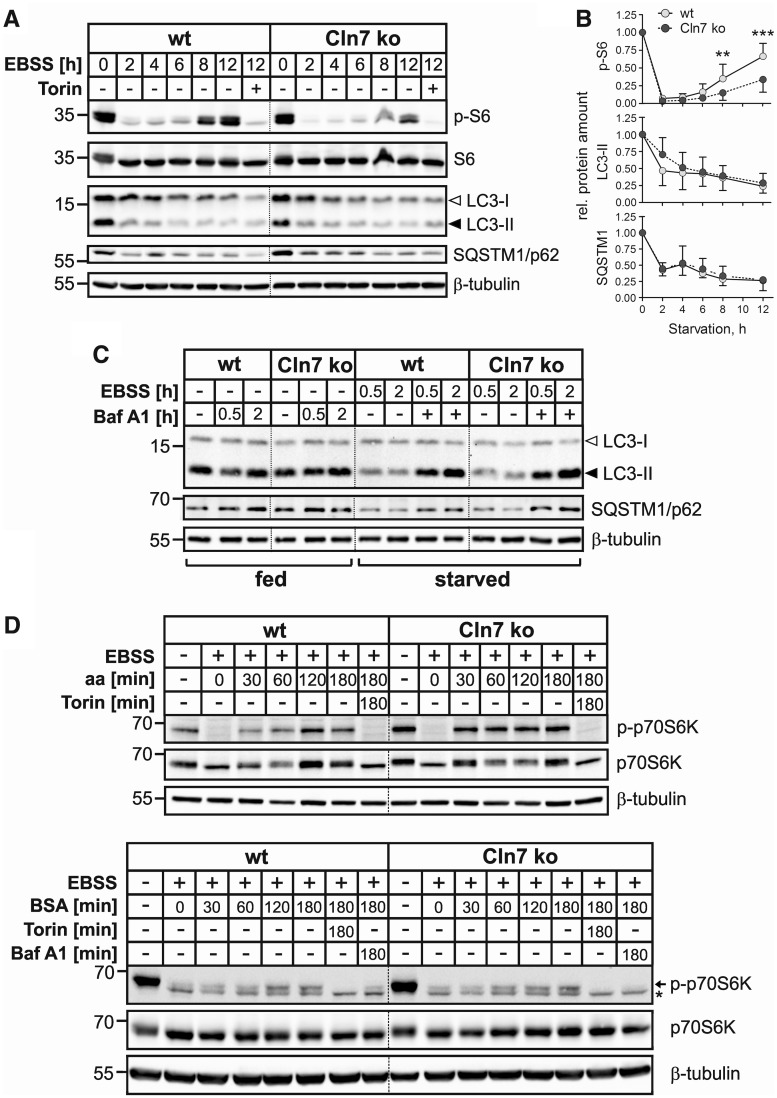
Defects in the MFSD8 gene encoding the lysosomal membrane protein CLN7 lead to CLN7 disease, a neurodegenerative lysosomal storage disorder belonging to the group of neuronal ceroid lipofuscinoses. Here, we have performed a SILAC-based quantitative analysis of the lysosomal proteome using Cln7-deficient mouse embryonic fibroblasts (MEFs) from a Cln7 knockout (ko) mouse model. From 3335 different proteins identified, we detected 56 soluble lysosomal proteins and 29 highly abundant lysosomal membrane proteins. Quantification revealed that the amounts of 12 different soluble lysosomal proteins were significantly reduced in Cln7 ko MEFs compared with wild-type controls. One of the most significantly depleted lysosomal proteins was Cln5 protein that underlies another distinct neuronal ceroid lipofuscinosis disorder. Expression analyses showed that the mRNA expression, biosynthesis, intracellular sorting and proteolytic processing of Cln5 were not affected, whereas the depletion of mature Cln5 protein was due to increased proteolytic degradation by cysteine proteases in Cln7 ko lysosomes. Considering the similar phenotypes of CLN5 and CLN7 patients, our data suggest that depletion of CLN5 may play an important part in the pathogenesis of CLN7 disease. In addition, we found a defect in the ability of Cln7 ko MEFs to adapt to starvation conditions as shown by impaired mammalian target of rapamycin complex 1 reactivation, reduced autolysosome tubulation and increased perinuclear accumulation of autolysosomes compared with controls. In summary, depletion of multiple soluble lysosomal proteins suggest a critical role of CLN7 for lysosomal function, which may contribute to the pathogenesis and progression of CLN7 disease.
Figures
Similar articles
-
Lysosomal dysfunction and impaired autophagy in a novel mouse model deficient for the lysosomal membrane protein Cln7.Hum Mol Genet. 2016 Feb 15;25(4):777-91. doi: 10.1093/hmg/ddv615. Epub 2015 Dec 17. Hum Mol Genet. 2016. PMID: 26681805
-
Gene disruption of Mfsd8 in mice provides the first animal model for CLN7 disease.Neurobiol Dis. 2014 May;65:12-24. doi: 10.1016/j.nbd.2014.01.003. Epub 2014 Jan 11. Neurobiol Dis. 2014. PMID: 24423645
-
A newly generated neuronal cell model of CLN7 disease reveals aberrant lysosome motility and impaired cell survival.Mol Genet Metab. 2019 Feb;126(2):196-205. doi: 10.1016/j.ymgme.2018.09.009. Epub 2018 Oct 1. Mol Genet Metab. 2019. PMID: 30301600
-
Molecular basis of the neuronal ceroid lipofuscinoses: mutations in CLN1, CLN2, CLN3, and CLN5.Hum Mutat. 1999;14(3):199-215. doi: 10.1002/(SICI)1098-1004(1999)14:3<199::AID-HUMU3>3.0.CO;2-A. Hum Mutat. 1999. PMID: 10477428 Review.
-
The intracellular location and function of proteins of neuronal ceroid lipofuscinoses.Brain Pathol. 2004 Jan;14(1):77-85. doi: 10.1111/j.1750-3639.2004.tb00501.x. Brain Pathol. 2004. PMID: 14997940 Free PMC article. Review.
Cited by
-
Lysosomal Proteomics Links Disturbances in Lipid Homeostasis and Sphingolipid Metabolism to CLN5 Disease.Cells. 2022 Jun 4;11(11):1840. doi: 10.3390/cells11111840. Cells. 2022. PMID: 35681535 Free PMC article.
-
Mfsd8 Modulates Growth and the Early Stages of Multicellular Development in Dictyostelium discoideum.Front Cell Dev Biol. 2022 Jun 9;10:930235. doi: 10.3389/fcell.2022.930235. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35756993 Free PMC article.
-
Cellular models of Batten disease.Biochim Biophys Acta Mol Basis Dis. 2020 Sep 1;1866(9):165559. doi: 10.1016/j.bbadis.2019.165559. Epub 2019 Oct 23. Biochim Biophys Acta Mol Basis Dis. 2020. PMID: 31655107 Free PMC article. Review.
-
Pathological Functions of Lysosomal Ion Channels in the Central Nervous System.Int J Mol Sci. 2024 Jun 14;25(12):6565. doi: 10.3390/ijms25126565. Int J Mol Sci. 2024. PMID: 38928271 Free PMC article. Review.
-
Recent Insight into the Genetic Basis, Clinical Features, and Diagnostic Methods for Neuronal Ceroid Lipofuscinosis.Int J Mol Sci. 2022 May 20;23(10):5729. doi: 10.3390/ijms23105729. Int J Mol Sci. 2022. PMID: 35628533 Free PMC article. Review.
References
-
- Mole S.E., Williams R.E., Goebel H.H. (2011) The Neuronal Ceroid Lipofuscinoses (Batten Disease) Contemporary Neurology Series, 2nd edn Oxford University Press Inc, New York.
-
- Kollmann K., Uusi-Rauva K., Scifo E., Tyynelä J., Jalanko A., Braulke T. (2013) Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim. Biophys. Acta, 1832, 1866–1881. - PubMed
-
- Kousi M., Lehesjoki A.E., Mole S.E. (2012) Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat., 33, 42–63. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
