

A VCP inhibitor substrate trapping approach (VISTA) enables proteomic profiling of endogenous ERAD substrates
- PMID: 29514927
- PMCID: PMC5921570
- DOI: 10.1091/mbc.E17-08-0514
A VCP inhibitor substrate trapping approach (VISTA) enables proteomic profiling of endogenous ERAD substrates
Abstract
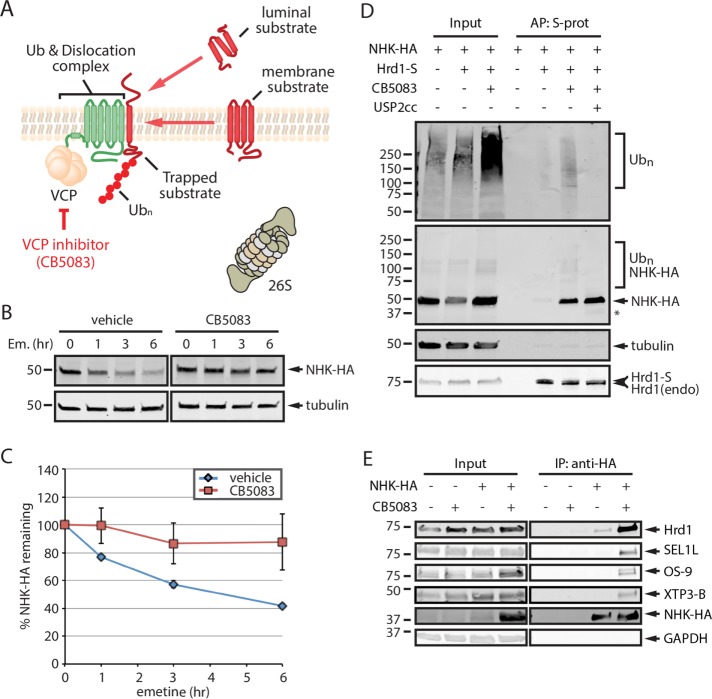
Endoplasmic reticulum (ER)-associated degradation (ERAD) mediates the proteasomal clearance of proteins from the early secretory pathway. In this process, ubiquitinated substrates are extracted from membrane-embedded dislocation complexes by the AAA ATPase VCP and targeted to the cytosolic 26S proteasome. In addition to its well-established role in the degradation of misfolded proteins, ERAD also regulates the abundance of key proteins such as enzymes involved in cholesterol synthesis. However, due to the lack of generalizable methods, our understanding of the scope of proteins targeted by ERAD remains limited. To overcome this obstacle, we developed a VCP inhibitor substrate trapping approach (VISTA) to identify endogenous ERAD substrates. VISTA exploits the small-molecule VCP inhibitor CB5083 to trap ERAD substrates in a membrane-associated, ubiquitinated form. This strategy, coupled with quantitative ubiquitin proteomics, identified previously validated (e.g., ApoB100, Insig2, and DHCR7) and novel (e.g., SCD1 and RNF5) ERAD substrates in cultured human hepatocellular carcinoma cells. Moreover, our results indicate that RNF5 autoubiquitination on multiple lysine residues targets it for ubiquitin and VCP--dependent clearance. Thus, VISTA provides a generalizable discovery method that expands the available toolbox of strategies to elucidate the ERAD substrate landscape.
Figures




References
-
- Bagola K, Mehnert M, Jarosch E, Sommer T. (2011). Protein dislocation from the ER. Biochim Biophys Acta , 925–936. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
