

Computational Strategies for Dissecting the High-Dimensional Complexity of Adaptive Immune Repertoires
- PMID: 29515569
- PMCID: PMC5826328
- DOI: 10.3389/fimmu.2018.00224
Computational Strategies for Dissecting the High-Dimensional Complexity of Adaptive Immune Repertoires
Abstract
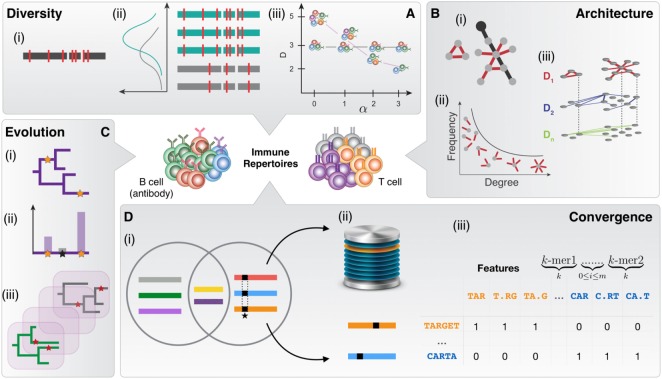
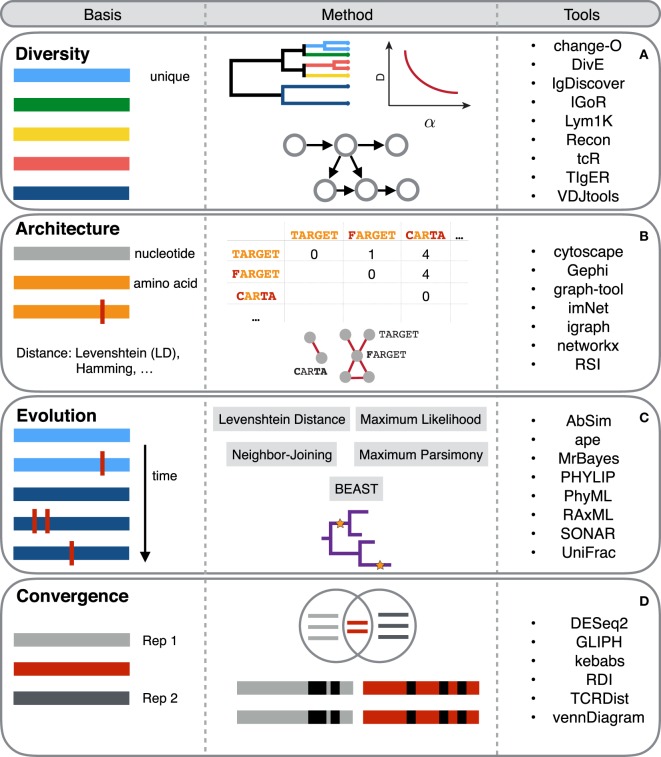
The adaptive immune system recognizes antigens via an immense array of antigen-binding antibodies and T-cell receptors, the immune repertoire. The interrogation of immune repertoires is of high relevance for understanding the adaptive immune response in disease and infection (e.g., autoimmunity, cancer, HIV). Adaptive immune receptor repertoire sequencing (AIRR-seq) has driven the quantitative and molecular-level profiling of immune repertoires, thereby revealing the high-dimensional complexity of the immune receptor sequence landscape. Several methods for the computational and statistical analysis of large-scale AIRR-seq data have been developed to resolve immune repertoire complexity and to understand the dynamics of adaptive immunity. Here, we review the current research on (i) diversity, (ii) clustering and network, (iii) phylogenetic, and (iv) machine learning methods applied to dissect, quantify, and compare the architecture, evolution, and specificity of immune repertoires. We summarize outstanding questions in computational immunology and propose future directions for systems immunology toward coupling AIRR-seq with the computational discovery of immunotherapeutics, vaccines, and immunodiagnostics.
Keywords: B-cell receptor; T-cell receptor; antibody discovery; artificial intelligence; immunogenomics; networks; phylogenetics; systems immunology.
Figures
Similar articles
-
Simulation of adaptive immune receptors and repertoires with complex immune information to guide the development and benchmarking of AIRR machine learning.Nucleic Acids Res. 2025 Jan 24;53(3):gkaf025. doi: 10.1093/nar/gkaf025. Nucleic Acids Res. 2025. PMID: 39873270 Free PMC article.
-
Bioinformatic and Statistical Analysis of Adaptive Immune Repertoires.Trends Immunol. 2015 Nov;36(11):738-749. doi: 10.1016/j.it.2015.09.006. Epub 2015 Oct 25. Trends Immunol. 2015. PMID: 26508293 Review.
-
Adaptive immune receptor repertoires, an overview of this exciting field.Immunol Lett. 2020 May;221:49-55. doi: 10.1016/j.imlet.2020.02.013. Epub 2020 Feb 27. Immunol Lett. 2020. PMID: 32113899 Review.
-
Adaptive Immune Receptor Repertoire (AIRR) Community Guide to TR and IG Gene Annotation.Methods Mol Biol. 2022;2453:279-296. doi: 10.1007/978-1-0716-2115-8_16. Methods Mol Biol. 2022. PMID: 35622332 Free PMC article.
-
AIRR Community Standardized Representations for Annotated Immune Repertoires.Front Immunol. 2018 Sep 28;9:2206. doi: 10.3389/fimmu.2018.02206. eCollection 2018. Front Immunol. 2018. PMID: 30323809 Free PMC article.
Cited by
-
Generation of a single-cell B cell atlas of antibody repertoires and transcriptomes to identify signatures associated with antigen specificity.iScience. 2023 Jan 25;26(3):106055. doi: 10.1016/j.isci.2023.106055. eCollection 2023 Mar 17. iScience. 2023. PMID: 36852274 Free PMC article.
-
Antigen-driven T cell responses in rheumatic diseases: insights from T cell receptor repertoire studies.Nat Rev Rheumatol. 2025 Mar;21(3):157-173. doi: 10.1038/s41584-025-01218-9. Epub 2025 Feb 7. Nat Rev Rheumatol. 2025. PMID: 39920282 Review.
-
Breast cancer is detectable from peripheral blood using machine learning over T cell receptor repertoires.NPJ Syst Biol Appl. 2025 Aug 8;11(1):89. doi: 10.1038/s41540-025-00573-3. NPJ Syst Biol Appl. 2025. PMID: 40781233 Free PMC article.
-
AIRRscape: An interactive tool for exploring B-cell receptor repertoires and antibody responses.PLoS Comput Biol. 2022 Sep 20;18(9):e1010052. doi: 10.1371/journal.pcbi.1010052. eCollection 2022 Sep. PLoS Comput Biol. 2022. PMID: 36126074 Free PMC article.
-
B cell tolerance and autoimmunity: Lessons from repertoires.J Exp Med. 2024 Sep 2;221(9):e20231314. doi: 10.1084/jem.20231314. Epub 2024 Aug 2. J Exp Med. 2024. PMID: 39093312 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources