

Review
doi: 10.1016/j.neuron.2018.01.029.
A Systems Neuroscience Approach to Migraine
Affiliations
- PMID: 29518355
- PMCID: PMC6402597
- DOI: 10.1016/j.neuron.2018.01.029
Item in Clipboard
Review
A Systems Neuroscience Approach to Migraine
Neuron.
.
Abstract
Migraine is an extremely common but poorly understood nervous system disorder. We conceptualize migraine as a disorder of sensory network gain and plasticity, and we propose that this framing makes it amenable to the tools of current systems neuroscience.
Keywords: allodynia; cortical spreading depression; gain; migraine; pain; photophobia; plasticity; systems neuroscience.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
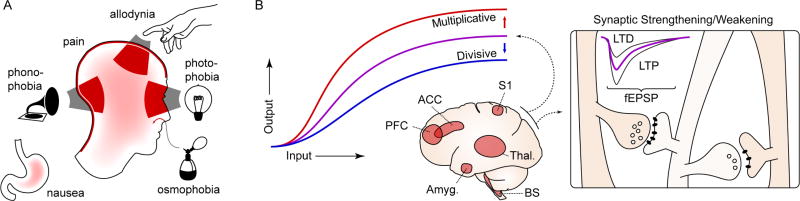
A. The migraine attack involves changes in multiple sensory percepts. Head and neck pain are the prototypic features, but migraine attacks are nearly always accompanied by some combination of photophobia, phonophobia, and/or allodynia (the perception of light, sound, and touch as painful, respectively). Osmophobia - a heightened, often aversive sensitivity to smell - is another frequent feature. Finally, nausea - an aversive viscerosensation - affects most migraineurs during an attack. These changes can become constant in chronic migraine. B. Conceptualizing migraine as a short- and long-term disruption of network gain. Widely distributed sensory, homeostatic/autonomic, and affective networks are likely involved. The amplitude (or salience) of percepts to the same stimulus is increased, potentially consistent with a change in circuit gain. These changes are temporary when associated with the migraine attack, but in chronic migraine they can be constant, suggesting entrainment of plasticity processes. PFC: prefrontal cortex; ACC: anterior cingulate cortex; S1: primary sensory cortex; Thal.: thalamus; Amyg.: amygdala; BS: brain stem nuclei; LTP, LTD: long term potentiation, depression; fEPSP: field excitatory postsynaptic potential.
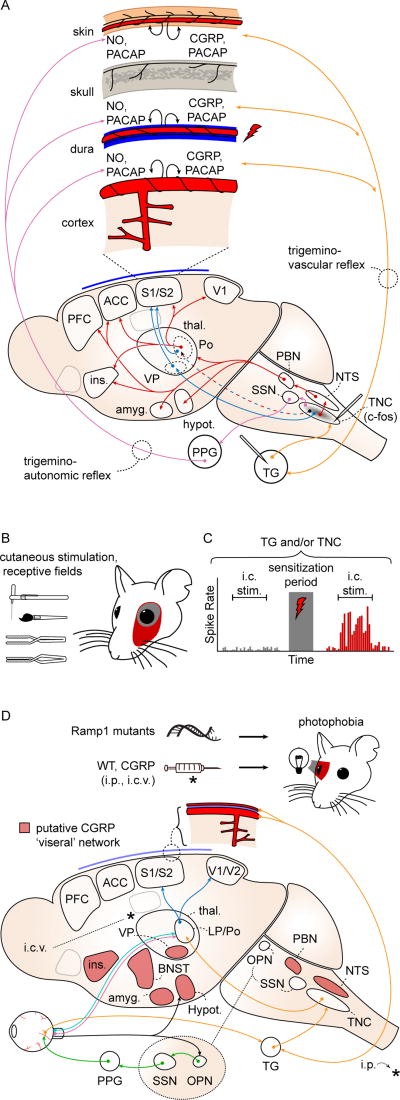
A. Craniofacial structures are innervated by nociceptors whose cell bodies are in the trigeminal ganglion (TG) and dorsal root ganglia of C1–3 (orange arrows; C1–3 not shown). The first central relay for craniofacial nociception is the trigeminal nucleus caudalis (TNC). TNC projects, directly and indirectly, to structures involved in the sensory/discriminative (blue arrows) and affective/motivational (red arrows) aspects of the pain percept. The trigeminovascular (orange arrows) and trigemino-autonomic (purple arrows) reflexes are activated by stimulation of nociceptive afferents (red lightning bolt), causing release of peptides and nitric oxide (black curved arrows) and generating a sustained nociceptive response that is used to model a migraine attack. ACC: anterior cingulate cortex; Amyg: amygdala; CGRP: calcitonin gene-related peptide; Hypot: hypothalamus; Ins: insula; NO: nitric oxide; NTS: nucleus tractus solitarius; PACAP: pituitary adenylate cyclase activated peptide; PBN: parabrachial nucleus; PFC: prefrontal cortex; Po: posterior nucleus of thalamus; S1/S2: primary, secondary somatosensory cortices; SSN: superior salivatory nucleus; Thal: thalamus; VP: ventroposterior nuclei of thalamus. B. Activation of craniofacial afferents results in persistent sensitization to sensory stimulation via von Frey filament, brush, pressure, or pinch; and expansion of cutaneous receptive fields (red) after application of intracranial inflammatory mediators. C. Schematized trigeminal ganglion or trigeminal nucleus caudalis neuron response to intracranial stimulation. Mechanosensive afferents show a potentiated response consistent with mechanical intracranial hypersensitivity after application of intracranial inflammatory mediators. D. Photophobia circuits. The photophobia that accompanies the migraine attack has been modeled in animals. Retinal ganglion cells project to the olivary pretectal nucleus (OPN; black arrows). OPN projections activate superior salivatory nucleus (SSN), which via the pterygopalatine ganglion (PPG; green), cause ocular vasodilation and activation of trigeminal afferents (orange) which densely innervate ocular blood vessels. These afferents, with cell bodies in the trigeminal ganglion, project to TNC, thalamus and cortex. Intrinsically photosensitive retinal ganglion cells (IPRGCs; pink) and cone cell-associated retinal ganglion cells (light blue) project directly to posterior thalamic neurons (primarily in lateral posterior and posterior nuclei; LP, Po) that also receive intracranial nociceptive afferents (orange). Retinal ganglion cells also project to hypothalamus (black). Posterior thalamic neurons fire in response to both light and pain stimuli, and their output projects diffusely to sensory and association cortices (dark blue; association cortex not shown). CGRP-overexpressing mice (Ramp1 mutants) are photophobic compared to littermates, and both intraperitoneal (i.p.) and intracerebroventricular (i.c.v.) CGRP injections cause photophobia in wild type mice. Red shading: putative CGRP 'visceral network.' BNST: bed nucleus of the stria terminalis. References in text.
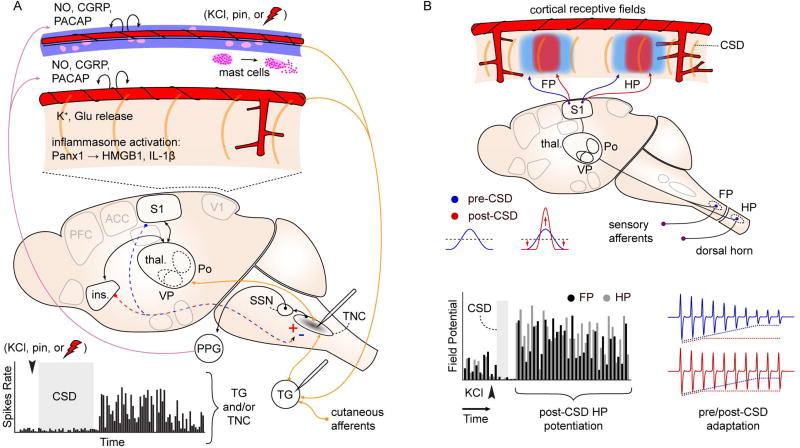
A. Spontaneous firing rate of TG and TNC neurons increases after experimentally-induced CSD (graph at bottom). Recording sites indicated on schematic. c-fos immediate early gene activation is also observed in TNC after CSD (gray shading). These changes are intepreted as consistent with activation of craniofacial nocieptive circuits. Schematic shows possible mechanisms: CSD is associated with release of K+ and glutamate, inflammosome activation (Panx1: neuronal Pannexin-1 receptor activation; HMGB1: high mobility group box 1; Il-1β; interleukin 1-beta), and mast cell degranulation, leading to activation of nociceptive afferents on pial and dural vessels. In addition to direct activation of peripheral nociceptive fibers, CSD can act through central, corticofugal pathways to affect evoked TNC firing. CSD in insula is associated with increased stimulus-evoked TNC firing, while CSD in somatosensory cortex is associated with a decrease (red/blue dashed arrows, +/−). This bi-directional modulation suggests complex effects of CSD on sensory networks. B. CSD alters cortical sensory mapping. Schematic shows sharpening of sensory map generated by potentiation at receptive field center and depression of response in surround regions. Graph: Summed evoked potential responses (sEP) are potentiated at receptive field center for up to 1.5 hrs after CSD passage. FP, HP: forepaw, hindpaw. Red,blue traces: Blunting of sensory adaptation after CSD. References in text.
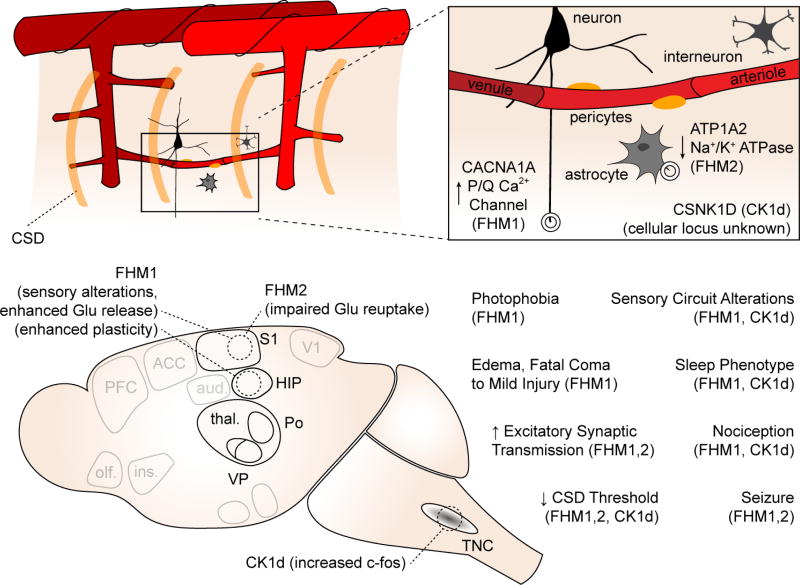
Top schematic shows genes involved and cellular loci (if known) of gene product. Bottom schematic shows key phenotypes observed in transgenic animals compared to wild type littermates.
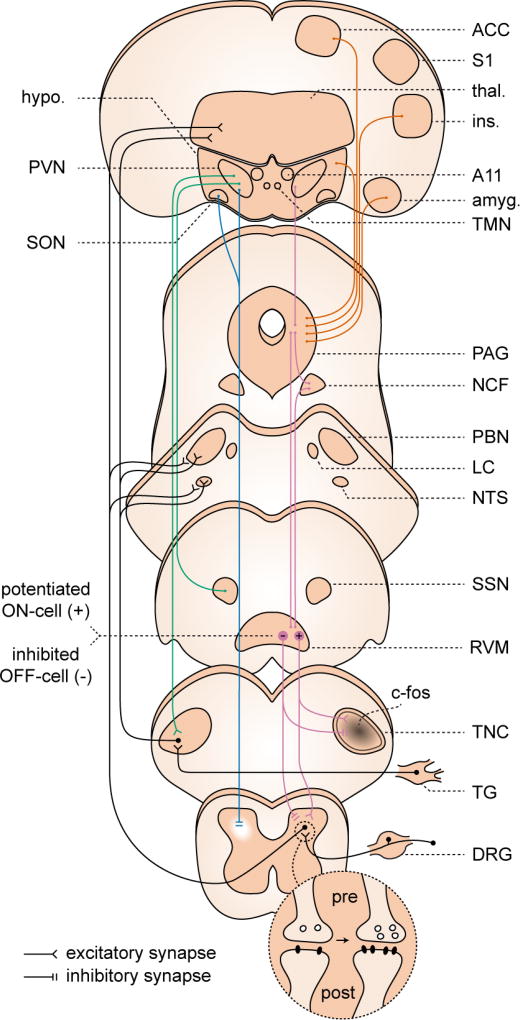
Dorsal horn and TNC. Both pre- and post-synaptic forms of LTP have been demonstrated in dorsal horn. These changes have not been conclusively demonstrated in TNC; however c-fos expression, which has been used as a proxy for LTP, has been observed in TNC to migraine-relevant stimuli. Descending modulation from periaqueductal gray (PAG), nucleus cuneiformis (NCF), and rostroventromedial medulla (RVM). Under cortical and hypothalamic control (orange traces) these structures contribute to widespread, bi-directional modulation of incoming signal from the dorsal horn (purple traces), and are implicated in sensitization in pain models. Potentiation (+) of RVM ON-cell and suppression (−) of OFF-cell activity has been observed in a migraine model. Hypothalamic descending modulation. There are direct projections from the paraventricular nucleus (PVN) of hypothalamus to the TNC, which increase TNC activation in a migraine model (green traces). More widespread projections from a small population of oxytocin-expressing (OT) neurons in PVN suppress dorsal horn wide dynamic range neuron firing (blue traces). This population is also responsible for reduction suppression of dorsal root ganglion (DRG) activity, via humoral release of OT (not shown). References in text.
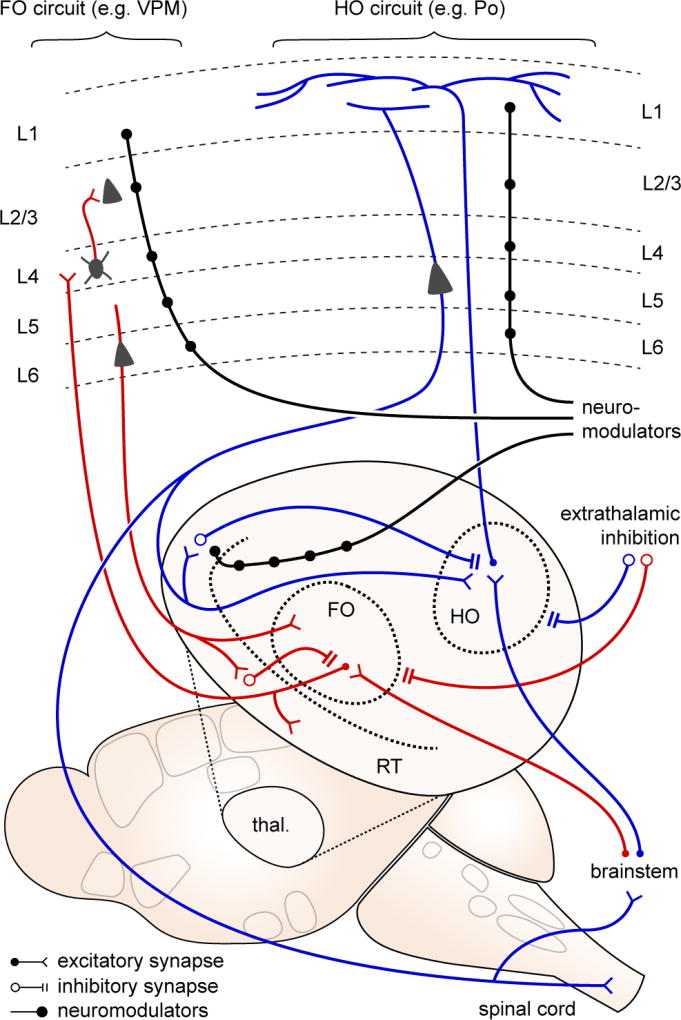
Thalamic relay nuclei are classified as first order (FO; red traces), where the primary (driver) input is from brainstem and spinal cord afferents; and higher order (HO; blue traces), where a significant proportion of driver input is from layer 5 of cortex. FO nuclei serve as sensory relays from the periphery, projecting primarily to cortical layer 4. HO nuclei may serve a more integrative role, projecting more diffusely thoughout the cortex, especially within layer 1. Thalamic activity is constrained by two sources of inhibition - local inhibition from the reticular nucleus (RT), and extrathalamic inhibition from zona incerta, basal ganglia, pretectal nucleus, and pontine reticular formation. ETI is suppressed in neuropathic pain models, leading to significantly increased activity in posterior (Po) thalamus, a HO relay. Both FO and HO circuits are susceptible to neuromodulation (black traces), at the thalamic and cortical levels.
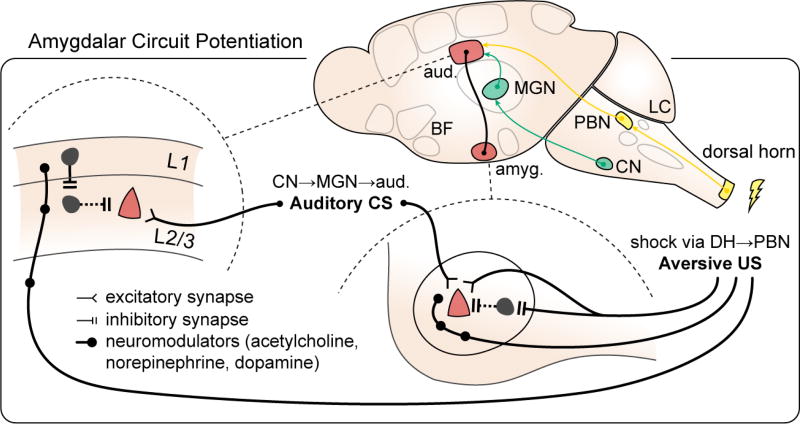
in fear learning paradigms. Fear learning provides a potentially migraine-relevant example of distributed circuit changes during an aversive event. Synaptic potentiation (red) occurs not only on pyramidal cell synapses in amygdala (right; lateral nucleus of amygdala shown) but also in the auditory cortex, the recipient region for the conditioned stimulus (CS, US: conditioned, unconditioned stimulus, pathways delineated in green and yellow, respectively; L1, L2/3: layers 1, 2/3; aud.: auditory cortex; amyg.: amygdala; CN: cochlear nucleus; MGN: medial geniculate nucleus; DH: dorsal horn; PBN: parabrachial nucleus; LC: locus ceruleus; BF: basal forebrain; adapted from Herry and Johansen, 2014).
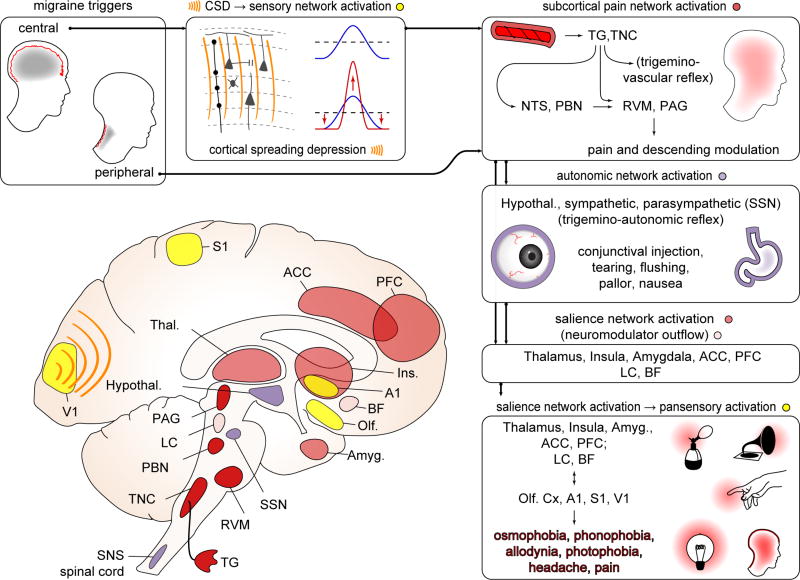
Boxes clockwise from top left, referring to diagram. Triggers can be central (e.g. CSD) or peripheral (e.g. NTG, CGRP infusion; unknown natural stimuli). Both central and peripheral triggers likely act through the common pathway of the craniofacial pain network (trigeminovascular system; see also Fig 2). CSD can trigger activation of trigeminal afferents. It can also directly modulate cortical network activity - for example through sensory map sharpening. In subcortical pain network activation (dark red structures), whether activated by CSD or peripheral triggers, trigeminal ganglion (TG) and trigeminal nucleus caudalis (TNC) neurons fire, leading to activation of higher pain/visceral network relays (nucleus tractus solitarius: NTS; parabrachial nucleus: PBN) and regions mediating descending modulation (periaqueductal gray: PAG; rostroventromedial medulla: RVM; locus ceruleus: LC). Depolarization of cranial nociceptive afferents can generate a pain percept, but the sustained pain of a migraine attack may require more extensive circuit activation (e.g. NTS, PBN and their multiple downstream structures), amplification (e.g. feed-forward effects of trigeminovascular reflex) and/or descending modulation (e.g. from PAG, RVM). Subcortical pain network activates autonomic centers: (purple structures) hypothalamus (Hypothal.), superior salivatory nucleus (SSN), LC, and brainstem and spinal cord sympathetic nuclei (SNS). SSN activation is likely responsible for conjunctival injection and tearing that can accompany migraine; broader autonomic network activation likely mediates nausea. Salience network activation (light red structures): Exteroceptive and interoceptive, first order and higher order nuclei in thalamus (Thal.) are activated, and relay to primary (S1) and secondary sensory cortices, insula (Ins.), and regions that encode the negative valence of pain: anterior cingulate cortex (ACC) and amygdala (Amyg.). Conscious awareness of headache likely requires activation of this distributed salience network. We hypothesize that this pain/salience network activation, likely in concert with neuromodulatory activity (pink structures; locus ceruleus (LC) and basal forebrain (BF) shown) is required for gain alteration in multiple sensory cortices (yellow structures) - primary visual cortex (V1) primary auditory cortex (A1), S1, olfactory cortex (Olf) - generating the sensory amplifications (photophobia, phonophobia, allodynia, osmophobia) that accompany the migraine attack. These amplifications, representing widely distributed cortical regions, are difficult to explain without network coordination.
References
-
- Alstadhaug KB. Migraine and the hypothalamus. Cephalalgia. 2009;29:809–17. - PubMed
-
- Ambrosini A, D’Alessio C, Magis D, Schoenen J. Targeting pericranial nerve branches to treat migraine: Current approaches and perspectives. Cephalalgia. 2015;35:1308–1322. - PubMed
-
- Ashina M, Hansen JM, À Dunga BO, Olesen J. Human models of migraine - short-term pain for long-term gain. Nat. Rev. Neurol. 2017;13:713–724. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
