

TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function
- PMID: 29518356
- PMCID: PMC5889092
- DOI: 10.1016/j.neuron.2018.01.031
TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function
Abstract
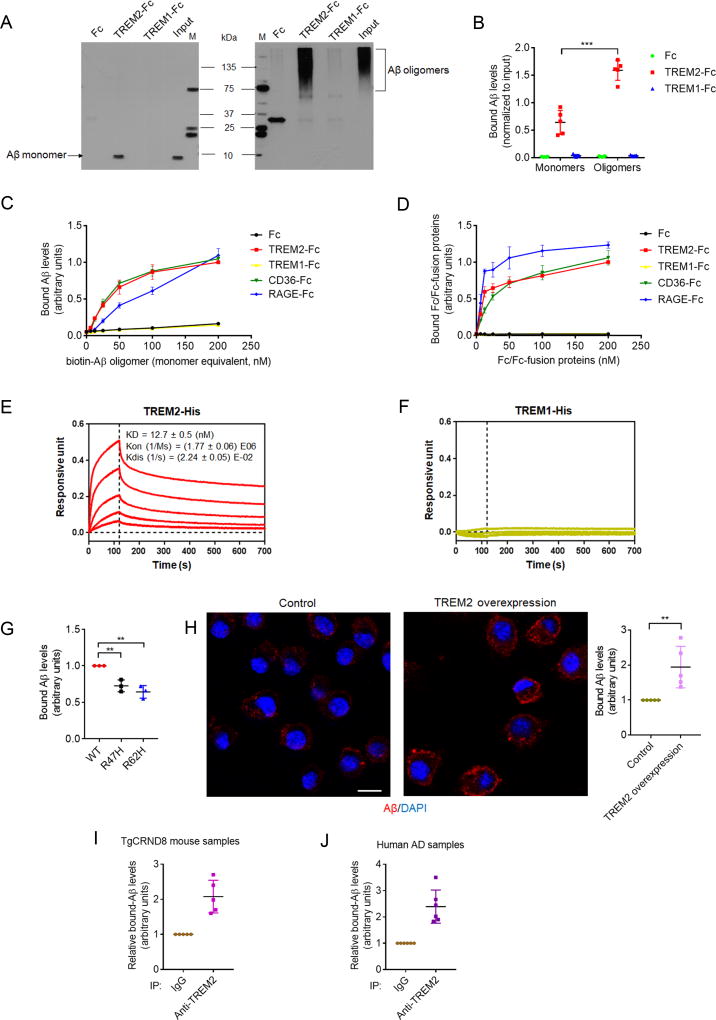
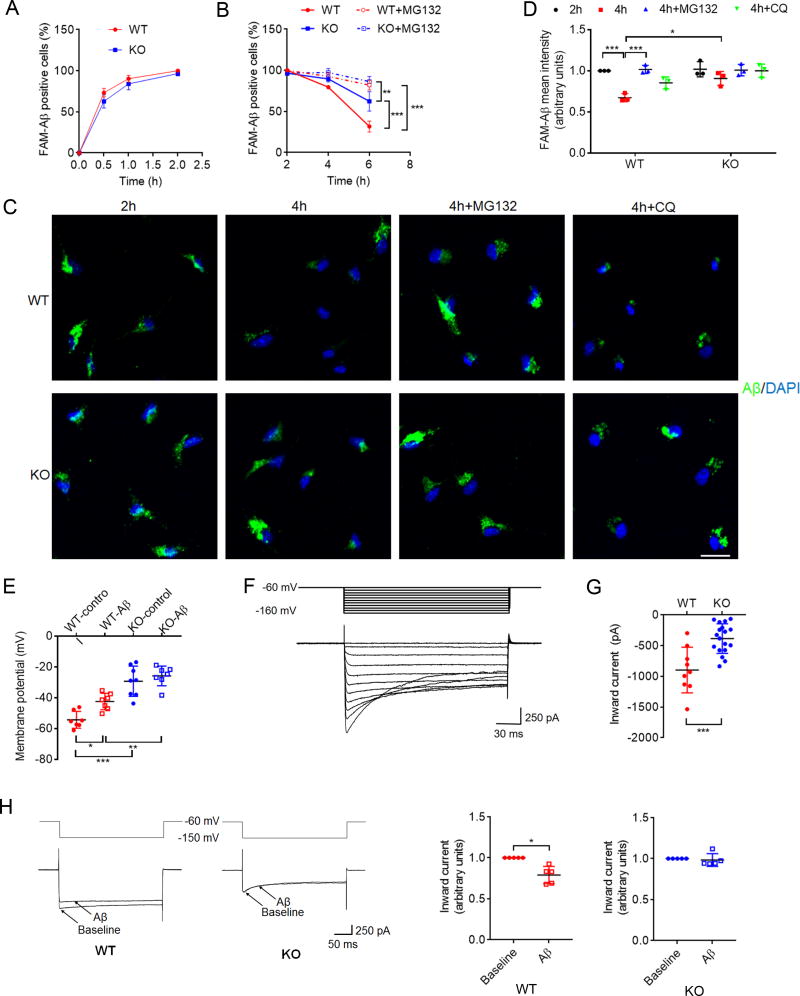
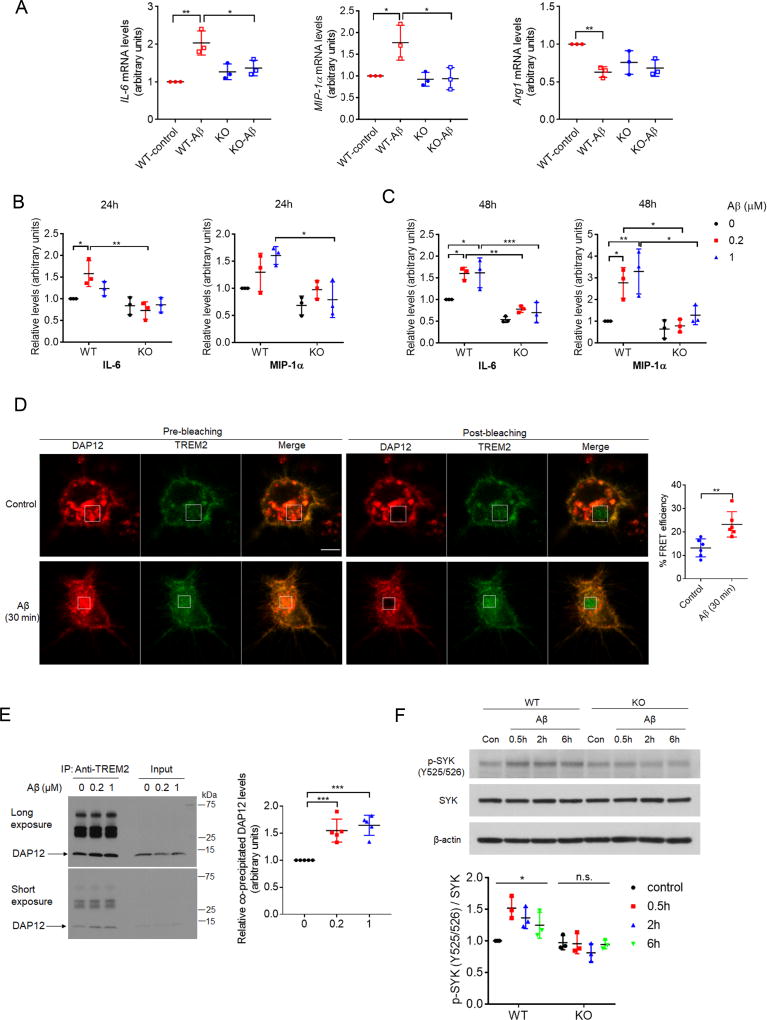
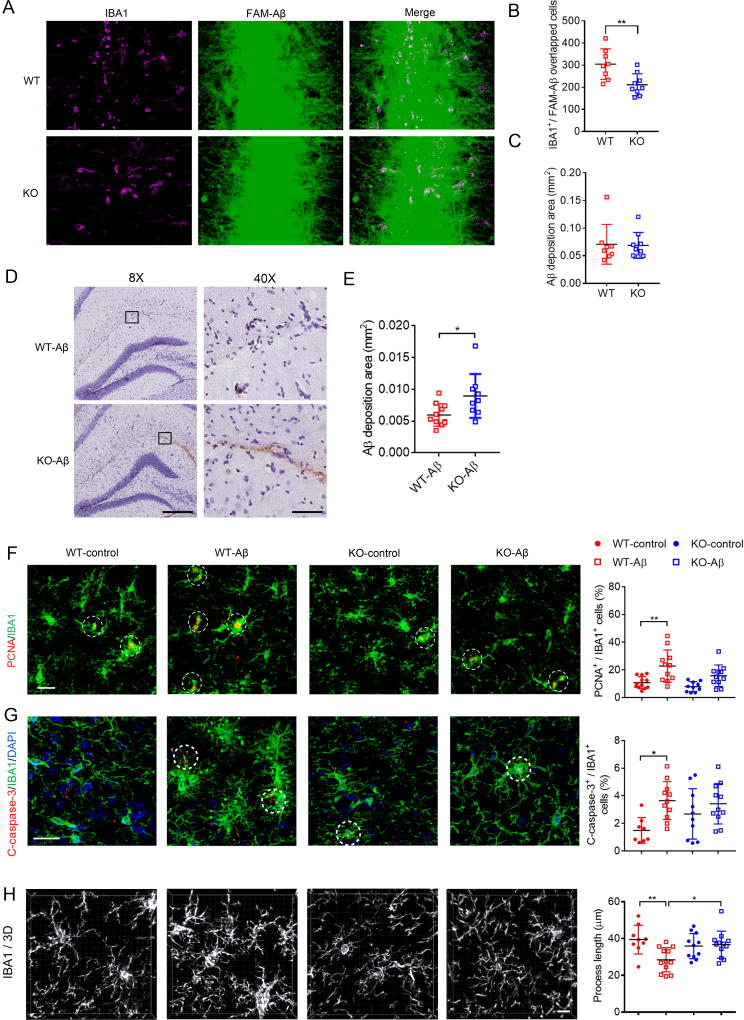
Mutations in triggering receptor expressed on myeloid cells 2 (TREM2) have been linked to increased Alzheimer's disease (AD) risk. Neurobiological functions of TREM2 and its pathophysiological ligands remain elusive. Here we found that TREM2 directly binds to β-amyloid (Aβ) oligomers with nanomolar affinity, whereas AD-associated TREM2 mutations reduce Aβ binding. TREM2 deficiency impairs Aβ degradation in primary microglial culture and mouse brain. Aβ-induced microglial depolarization, K+ inward current induction, cytokine expression and secretion, migration, proliferation, apoptosis, and morphological changes are dependent on TREM2. In addition, TREM2 interaction with its signaling adaptor DAP12 is enhanced by Aβ, regulating downstream phosphorylation of SYK and GSK3β. Our data demonstrate TREM2 as a microglial Aβ receptor transducing physiological and AD-related pathological effects associated with Aβ.
Keywords: Alzheimer’s disease; Aβ; R47H; TREM2; microglia; neuroinflammation.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
TREM2 and Amyloid Beta: A Love-Hate Relationship.Neuron. 2018 Mar 7;97(5):991-993. doi: 10.1016/j.neuron.2018.02.018. Neuron. 2018. PMID: 29518360
References
-
- Boucsein C, Kettenmann H, Nolte C. Electrophysiological properties of microglial cells in normal and pathologic rat brain slices. Eur J Neurosci. 2000;12:2049–2058. - PubMed
-
- Cavanaugh SE, Pippin JJ, Barnard ND. Animal models of Alzheimer disease: historical pitfalls and a path forward. ALTEX. 2014;31:279–302. - PubMed
-
- Chen J, Carey K, Godowski PJ. Identification of Gas6 as a ligand for Mer, a neural cell adhesion molecule related receptor tyrosine kinase implicated in cellular transformation. Oncogene. 1997;14:2033–2039. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
