

Fluvastatin inhibits advanced glycation end products-induced proliferation, migration, and extracellular matrix accumulation in vascular smooth muscle cells by targeting connective tissue growth factor
- PMID: 29520172
- PMCID: PMC5840078
- DOI: 10.4196/kjpp.2018.22.2.193
Fluvastatin inhibits advanced glycation end products-induced proliferation, migration, and extracellular matrix accumulation in vascular smooth muscle cells by targeting connective tissue growth factor
Abstract
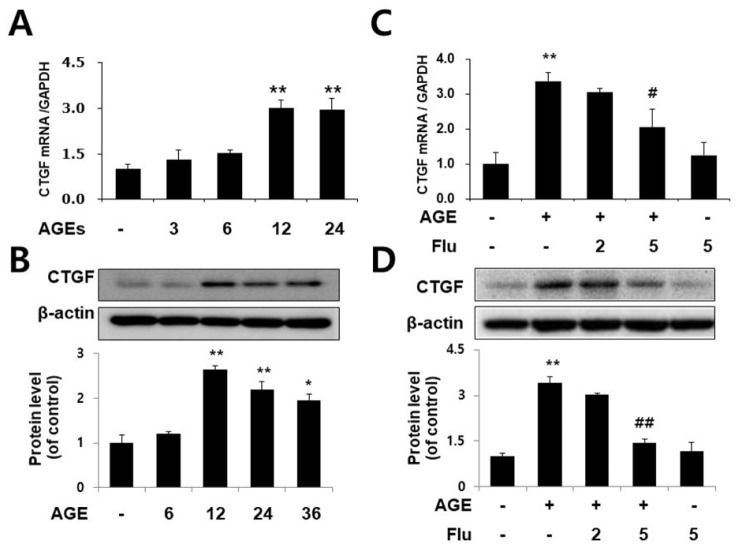
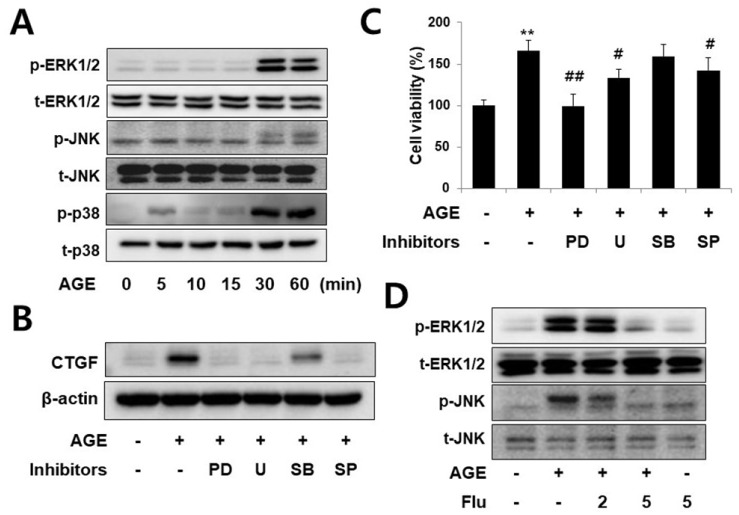
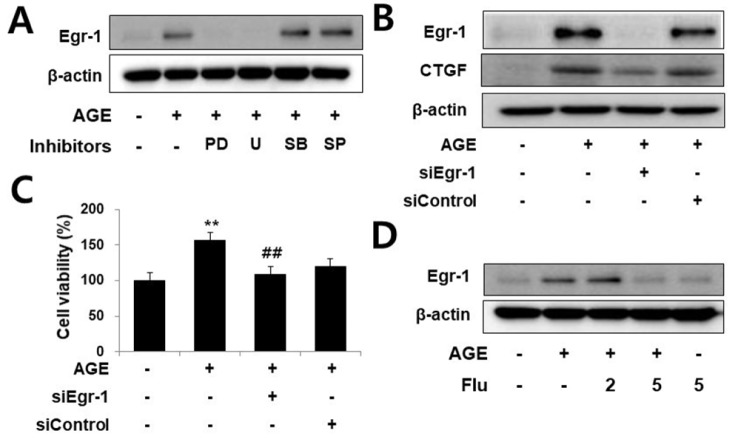
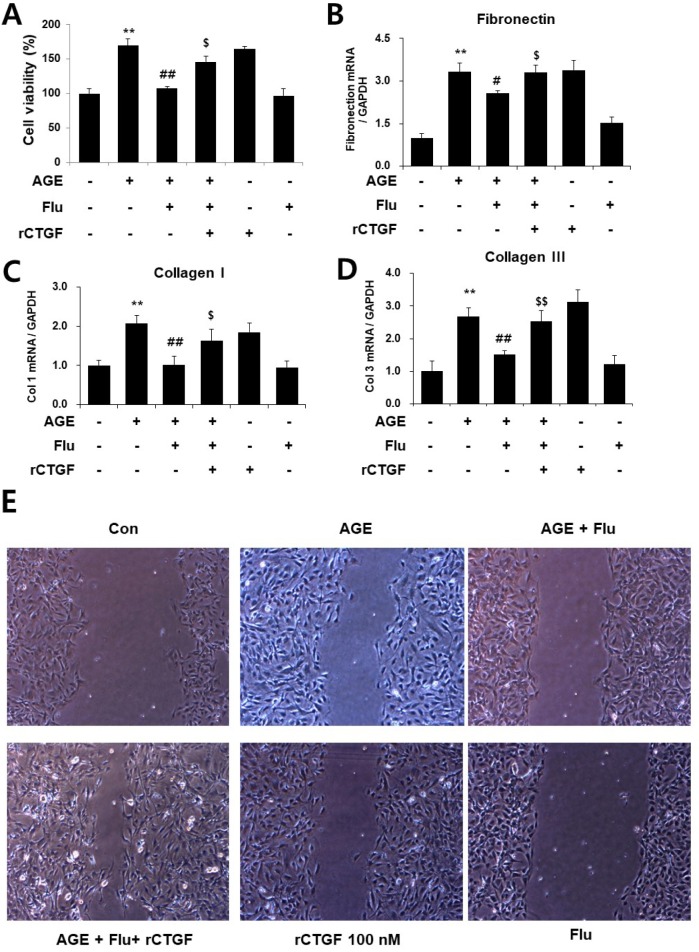
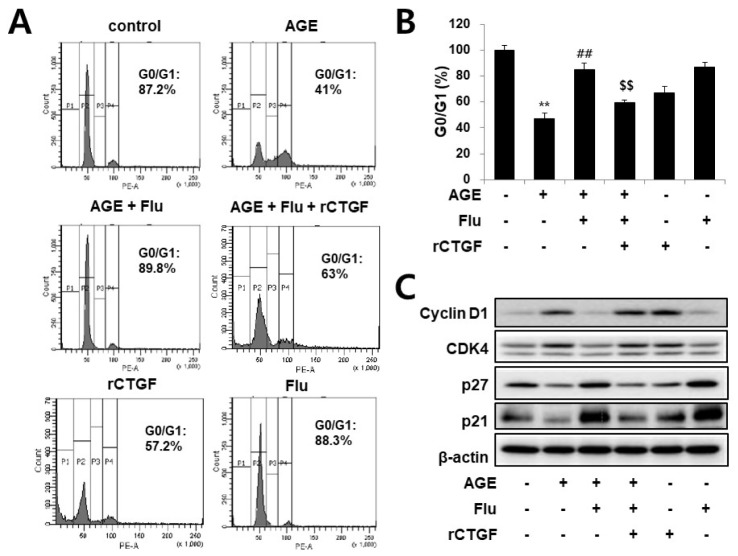
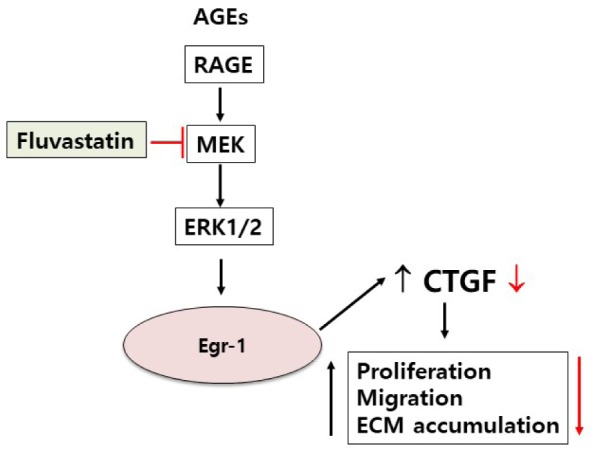
Connective tissue growth factor (CTGF) is a novel fibrotic mediator, which is considered to mediate fibrosis through extracellular matrix (ECM) synthesis in diabetic cardiovascular complications. Statins have significant immunomodulatory effects and reduce vascular injury. We therefore examined whether fluvastatin has anti-fibrotic effects in vascular smooth muscle cells (VSMCs) and elucidated its putative transduction signals. We show that advanced glycation end products (AGEs) stimulated CTGF mRNA and protein expression in a time-dependent manner. AGE-induced CTGF expression was mediated via ERK1/2, JNK, and Egr-1 pathways, but not p38; consequently, cell proliferation and migration and ECM accumulation were regulated by CTGF signaling pathway. AGE-stimulated VSMC proliferation, migration, and ECM accumulation were blocked by fluvastatin. However, the inhibitory effect of fluvastatin was restored by administration of CTGF recombinant protein. AGE-induced VSMC proliferation was dependent on cell cycle arrest, thereby increasing G1/G0 phase. Fluvastatin repressed cell cycle regulatory genes cyclin D1 and Cdk4 and augmented cyclin-dependent kinase inhibitors p27 and p21 in AGE-induced VSMCs. Taken together, fluvastatin suppressed AGE-induced VSMC proliferation, migration, and ECM accumulation by targeting CTGF signaling mechanism. These findings might be evidence for CTGF as a potential therapeutic target in diabetic vasculature complication.
Keywords: Advanced glycation end products; Cell cycle arrest; Connective tissue growth factor; Extracellular matrix; Fluvastatin; Vascular smooth muscle cell.
Conflict of interest statement
CONFLICTS OF INTEREST: The authors declare no conflicts of interest.
Figures
References
-
- Makita Z, Bucala R, Rayfield EJ, Friedman EA, Kaufman AM, Korbet SM, Barth RH, Winston JA, Fuh H, Manogue KR, et al. Reactive glycosylation endproducts in diabetic uraemia and treatment of renal failure. Lancet. 1994;343:1519–1522. - PubMed
-
- Cooper ME. Importance of advanced glycation end products in diabetes-associated cardiovascular and renal disease. Am J Hypertens. 2004;17:31S–38S. - PubMed
-
- Reddy GK. AGE-related cross-linking of collagen is associated with aortic wall matrix stiffness in the pathogenesis of drug-induced diabetes in rats. Microvasc Res. 2004;68:132–142. - PubMed
-
- Throckmorton DC, Brogden AP, Min B, Rasmussen H, Kashgarian M. PDGF and TGF-beta mediate collagen production by mesangial cells exposed to advanced glycosylation end products. Kidney Int. 1995;48:111–117. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
