

Adrenomedullin and Adrenomedullin-Targeted Therapy As Treatment Strategies Relevant for Sepsis
- PMID: 29520277
- PMCID: PMC5827550
- DOI: 10.3389/fimmu.2018.00292
Adrenomedullin and Adrenomedullin-Targeted Therapy As Treatment Strategies Relevant for Sepsis
Abstract
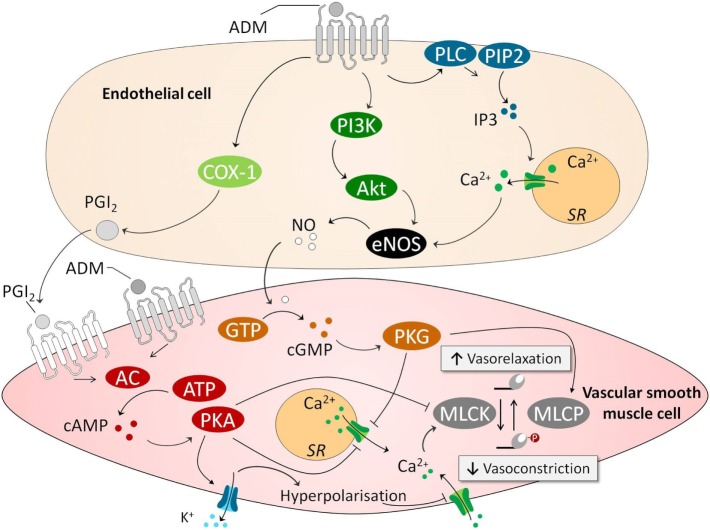
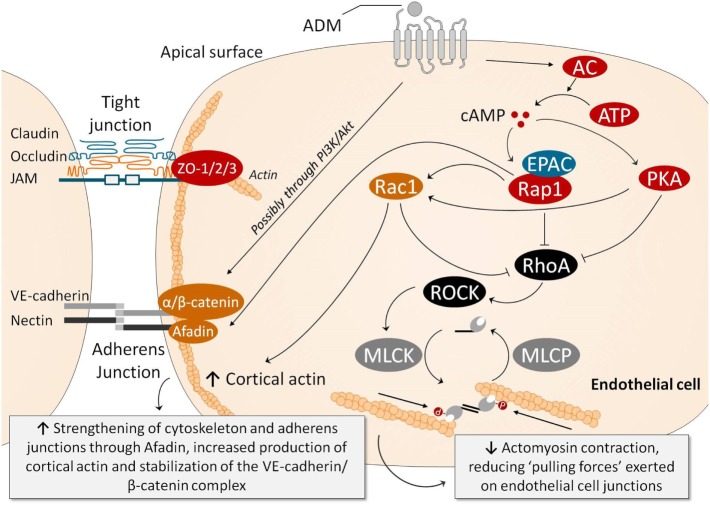
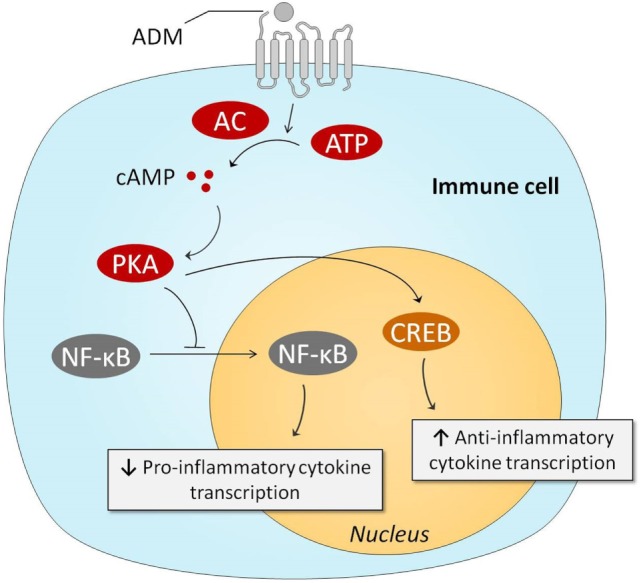
Sepsis remains a major medical challenge, for which, apart from improvements in supportive care, treatment has not relevantly changed over the last few decades. Vasodilation and vascular leakage play a pivotal role in the development of septic shock, with vascular leakage being caused by disrupted endothelial integrity. Adrenomedullin (ADM), a free circulating peptide involved in regulation of endothelial barrier function and vascular tone, is implicated in the pathophysiology of sepsis. ADM levels are increased during sepsis, and correlate with extent of vasodilation, as well as with disease severity and mortality. In vitro and preclinical in vivo data show that administration of ADM exerts anti-inflammatory, antimicrobial, and protective effects on endothelial barrier function during sepsis, but other work suggests that it may also decrease blood pressure, which could be detrimental for patients with septic shock. Work has been carried out to negate ADMs putative negative effects, while preserving or even potentiating its beneficial actions. Preclinical studies have demonstrated that the use of antibodies that bind to the N-terminus of ADM results in an overall increase of circulating ADM levels and improves sepsis outcome. Similar beneficial effects were obtained using coadministration of ADM and ADM-binding protein-1. It is hypothesized that the mechanism behind the beneficial effects of ADM binding involves prolongation of its half-life and a shift of ADM from the interstitium to the circulation. This in turn results in increased ADM activity in the blood compartment, where it exerts beneficial endothelial barrier-stabilizing effects, whereas its detrimental vasodilatory effects in the interstitium are reduced. Up till now, in vivo data on ADM-targeted treatments in humans are lacking; however, the first study in septic patients with an N-terminus antibody (Adrecizumab) is currently being conducted.
Keywords: adrenomedullin; antibodies; sepsis; septic shock; treatment; vascular barrier function.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials