

Accurate, Sensitive, and Precise Multiplexed Proteomics Using the Complement Reporter Ion Cluster
- PMID: 29522331
- PMCID: PMC6220677
- DOI: 10.1021/acs.analchem.7b04713
Accurate, Sensitive, and Precise Multiplexed Proteomics Using the Complement Reporter Ion Cluster
Abstract
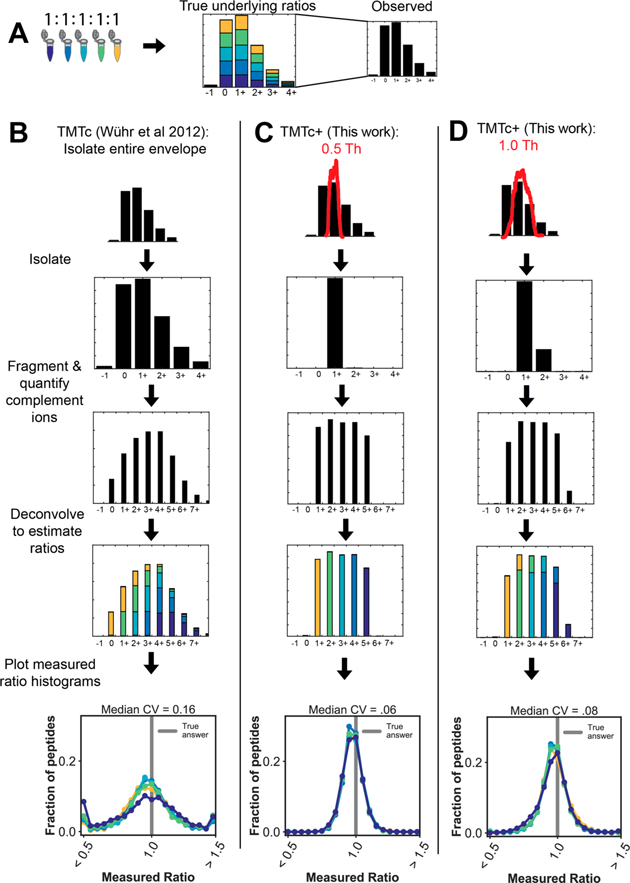
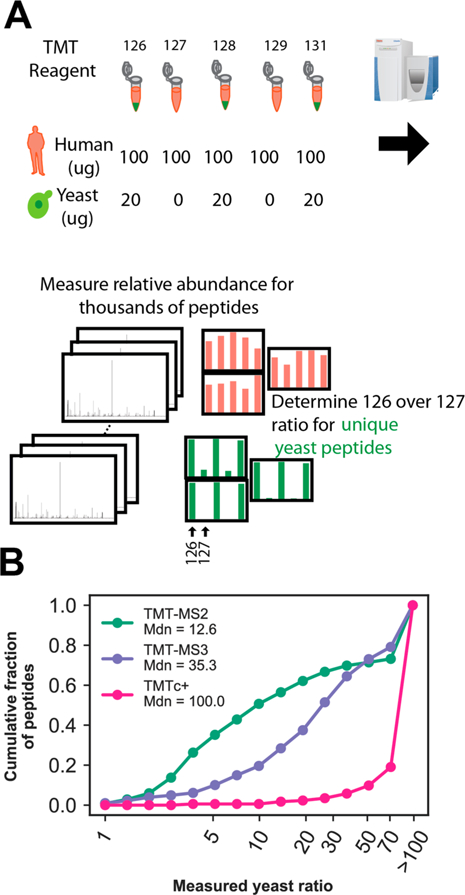
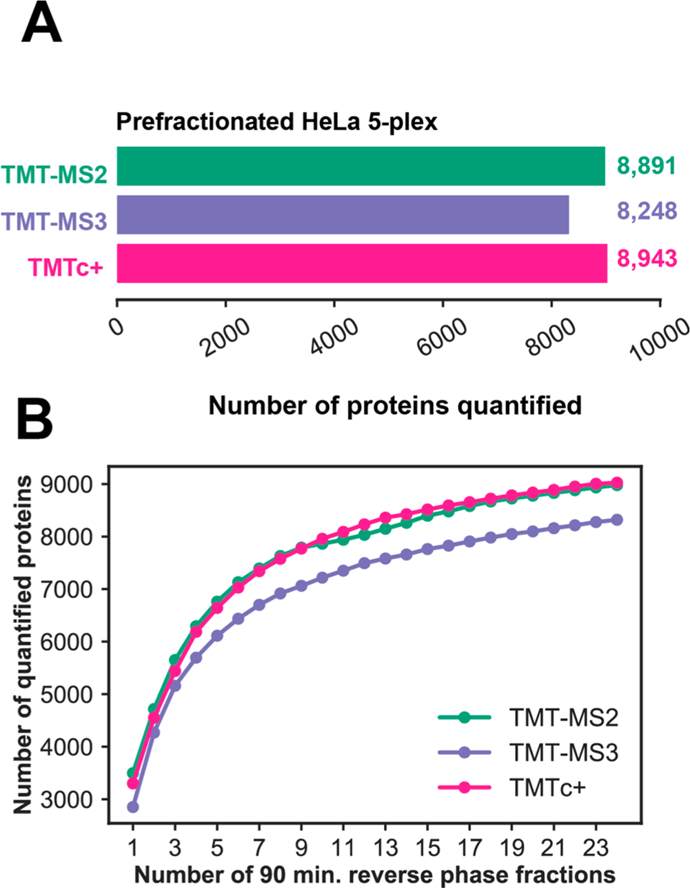
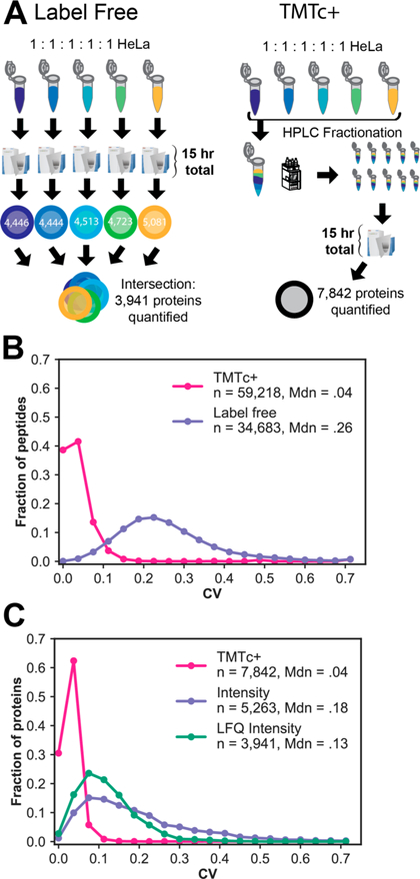
Quantitative analysis of proteomes across multiple time points, organelles, and perturbations is essential for understanding both fundamental biology and disease states. The development of isobaric tags (e.g., TMT) has enabled the simultaneous measurement of peptide abundances across several different conditions. These multiplexed approaches are promising in principle because of advantages in throughput and measurement quality. However, in practice, existing multiplexing approaches suffer from key limitations. In its simple implementation (TMT-MS2), measurements are distorted by chemical noise leading to poor measurement accuracy. The current state-of-the-art (TMT-MS3) addresses this but requires specialized quadrupole-iontrap-Orbitrap instrumentation. The complement reporter ion approach (TMTc) produces high accuracy measurements and is compatible with many more instruments, like quadrupole-Orbitraps. However, the required deconvolution of the TMTc cluster leads to poor measurement precision. Here, we introduce TMTc+, which adds the modeling of the MS2-isolation step into the deconvolution algorithm. The resulting measurements are comparable in precision to TMT-MS3/MS2. The improved duty cycle and lower filtering requirements make TMTc+ more sensitive than TMT-MS3 and comparable with TMT-MS2. At the same time, unlike TMT-MS2, TMTc+ is exquisitely able to distinguish signal from chemical noise even outperforming TMT-MS3. Lastly, we compare TMTc+ to quantitative label-free proteomics of total HeLa lysate and find that TMTc+ quantifies 7.8k versus 3.9k proteins in a 5-plex sample. At the same time, the median coefficient of variation improves from 13% to 4%. Thus, TMTc+ advances quantitative proteomics by enabling accurate, sensitive, and precise multiplexed experiments on more commonly used instruments.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Thompson A; Schafer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Anal. Chem 2003, 75, 1895–1904. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
