

Prematurity, ventricular septal defect and dysmorphisms are independent predictors of pathogenic copy number variants: a retrospective study on array-CGH results and phenotypical features of 293 children with neurodevelopmental disorders and/or multiple congenital anomalies
- PMID: 29523172
- PMCID: PMC5845186
- DOI: 10.1186/s13052-018-0467-z
Prematurity, ventricular septal defect and dysmorphisms are independent predictors of pathogenic copy number variants: a retrospective study on array-CGH results and phenotypical features of 293 children with neurodevelopmental disorders and/or multiple congenital anomalies
Abstract
Background: Since 2010, array-CGH (aCGH) has been the first-tier test in the diagnostic approach of children with neurodevelopmental disorders (NDD) or multiple congenital anomalies (MCA) of unknown origin. Its broad application led to the detection of numerous variants of uncertain clinical significance (VOUS). How to appropriately interpret aCGH results represents a challenge for the clinician.
Method: We present a retrospective study on 293 patients with age range 1 month - 29 years (median 7 years) with NDD and/or MCA and/or dysmorphisms, investigated through aCGH between 2005 and 2016. The aim of the study was to analyze clinical and molecular cytogenetic data in order to identify what elements could be useful to interpret unknown or poorly described aberrations. Comparison of phenotype and cytogenetic characteristics through univariate analysis and multivariate logistic regression was performed.
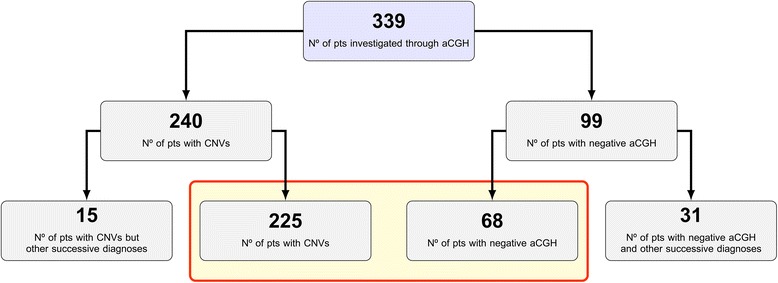
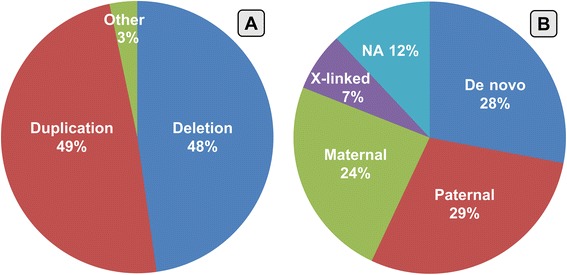
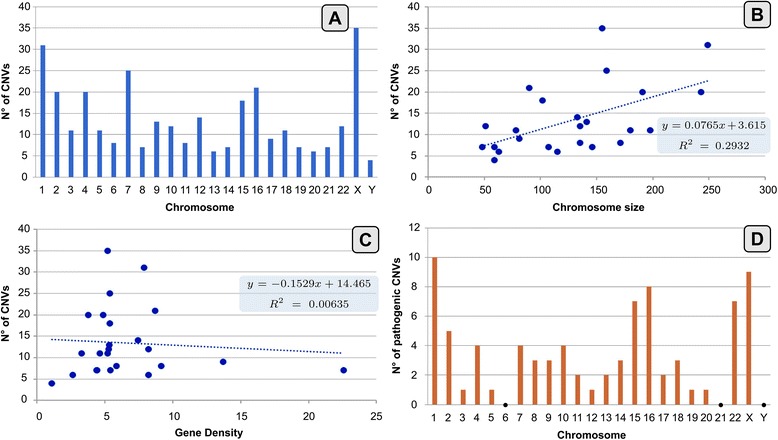
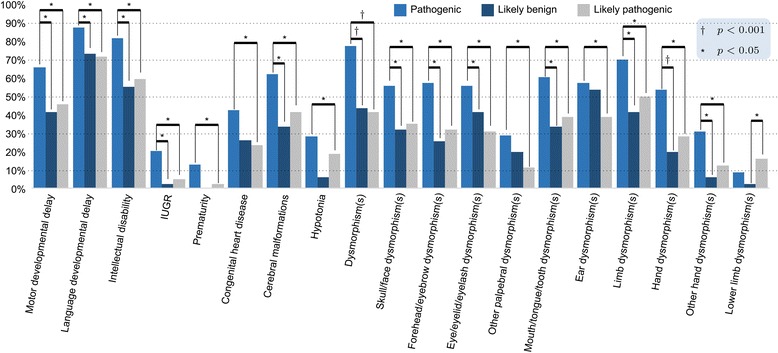
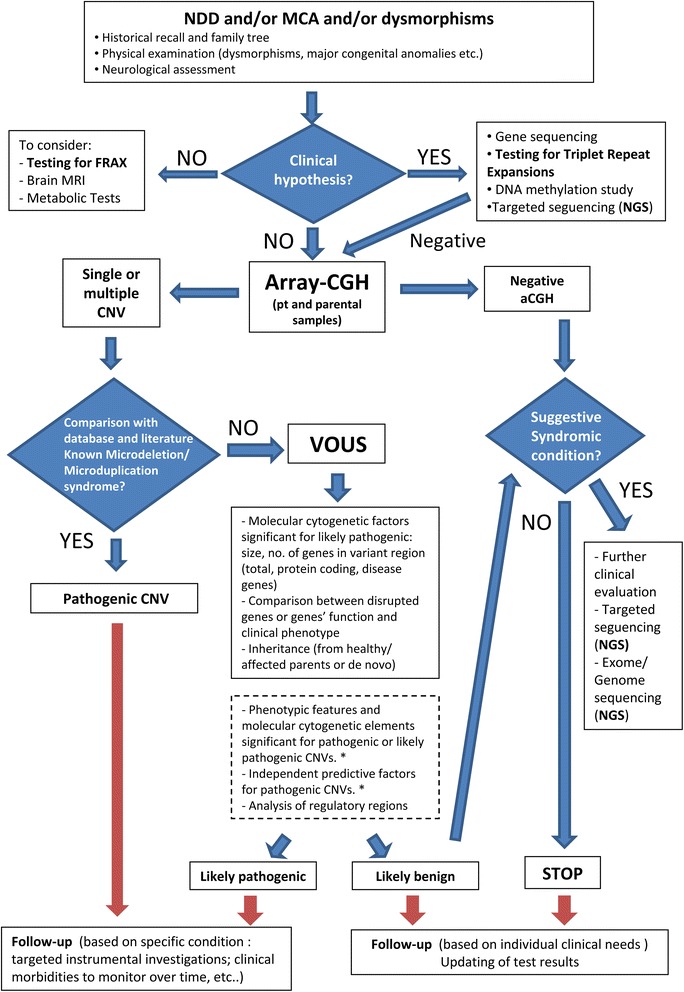
Results: Copy number variations (CNVs) with a frequency < 1% were detected in 225 patients of the total sample, while 68 patients presented only variants with higher frequency (heterozygous deletions or amplification) and were considered to have negative aCGH. Proved pathogenic CNVs were detected in 70 patients (20.6%). Delayed psychomotor development, intellectual disability, intrauterine growth retardation (IUGR), prematurity, congenital heart disease, cerebral malformations and dysmorphisms correlated to reported pathogenic CNVs. Prematurity, ventricular septal defect and dysmorphisms remained significant predictors of pathogenic CNVs in the multivariate logistic model whereas abnormal EEG and limb dysmorphisms were mainly detected in the group with likely pathogenic VOUS. A flow-chart regarding the care for patients with NDD and/or MCA and/or dysmorphisms and the interpretation of aCGH has been made on the basis of the data inferred from this study and literature.
Conclusion: Our work contributes to make the investigative process of CNVs more informative and suggests possible directions in aCGH interpretation and phenotype correlation.
Keywords: Array-CGH; Dysmorphisms; Interpretation; Multiple congenital anomalies; Neurodevelopmental disorders.
Conflict of interest statement
Ethics approval and consent to participate
According to the national laws, ethics approval by the locally appointed Ethics Committee was not required because: no intervention was applied; no identifiable private information was collected; patients underwent only routine diagnostic procedures, according to current guidelines of the Italian Society of Human Genetics; and an anonymized dataset was analysed.
Consent for publication
Not applicable.
This is a retrospective, observational study. At the time when the patients were clinically examined and molecular tests were carried out, the following elements of consent were required of the parents: consent to have personal data dealt with, consent to have genetic data treated, consent to research, consent to the execution of genetic test, on condition that the parents received an information sheet on the genetic test and a verbal explanation.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
New insights in the interpretation of array-CGH: autism spectrum disorder and positive family history for intellectual disability predict the detection of pathogenic variants.Ital J Pediatr. 2016 Apr 12;42:39. doi: 10.1186/s13052-016-0246-7. Ital J Pediatr. 2016. PMID: 27072107 Free PMC article.
-
The clinical benefit of array-based comparative genomic hybridization for detection of copy number variants in Czech children with intellectual disability and developmental delay.BMC Med Genomics. 2019 Jul 23;12(1):111. doi: 10.1186/s12920-019-0559-7. BMC Med Genomics. 2019. PMID: 31337399 Free PMC article.
-
Predictive diagnostic value for the clinical features accompanying intellectual disability in children with pathogenic copy number variations: a multivariate analysis.Ital J Pediatr. 2014 Apr 28;40:39. doi: 10.1186/1824-7288-40-39. Ital J Pediatr. 2014. PMID: 24775911 Free PMC article.
-
Chromosome copy number variants in fetuses with syndromic malformations.Birth Defects Res. 2017 Jun 1;109(10):725-733. doi: 10.1002/bdr2.1054. Birth Defects Res. 2017. PMID: 28568742 Review.
-
Phenotype to genotype characterization by array-comparative genomic hydridization (a-CGH) in case of fetal malformations: A systematic review.Taiwan J Obstet Gynecol. 2019 Jan;58(1):15-28. doi: 10.1016/j.tjog.2018.11.003. Taiwan J Obstet Gynecol. 2019. PMID: 30638470
Cited by
-
Testing single/combined clinical categories on 5110 Italian patients with developmental phenotypes to improve array-based detection rate.Mol Genet Genomic Med. 2020 Jan;8(1):e1056. doi: 10.1002/mgg3.1056. Epub 2019 Dec 18. Mol Genet Genomic Med. 2020. PMID: 31851782 Free PMC article.
-
Rare Copy Number Variations and Predictors in Children With Intellectual Disability and Epilepsy.Front Neurol. 2018 Nov 19;9:947. doi: 10.3389/fneur.2018.00947. eCollection 2018. Front Neurol. 2018. PMID: 30510536 Free PMC article.
-
The diagnostic yield of molecular karyotyping: a retrospective single-center study.Croat Med J. 2025 May 7;66(2):92-99. doi: 10.3325/cmj.2025.66.92. Croat Med J. 2025. PMID: 40343432 Free PMC article.
-
Contribution of Congenital Heart Disorders Associated With Copy Number Variants in Mediating Risk for Brain Developmental Disorders: Evidence From 20-Year Retrospective Cohort Study.Front Cardiovasc Med. 2021 Jul 15;8:655463. doi: 10.3389/fcvm.2021.655463. eCollection 2021. Front Cardiovasc Med. 2021. PMID: 34336942 Free PMC article.
References
-
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. - DOI - PMC - PubMed
-
- Chong WW, Lo IF, Lam ST, Wang CC, Luk HM, Leung TY, et al. Performance of chromosomal microarray for patients with intellectual disabilities/developmental delay, autism, and multiple congenital anomalies in a Chinese cohort. Mol Cytogenet. 2014;7:34. doi: 10.1186/1755-8166-7-34. - DOI - PMC - PubMed
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
