

High-Throughput Analysis of DNA Break-Induced Chromosome Rearrangements by Amplicon Sequencing
- PMID: 29523230
- PMCID: PMC9703968
- DOI: 10.1016/bs.mie.2017.11.028
High-Throughput Analysis of DNA Break-Induced Chromosome Rearrangements by Amplicon Sequencing
Abstract
The mechanistic understanding of how DNA double-strand breaks (DSB) are repaired is rapidly advancing in part due to the advent of inducible site-specific break model systems as well as the employment of next-generation sequencing (NGS) technologies to sequence repair junctions at high depth. Unfortunately, the sheer volume of data produced by these methods makes it difficult to analyze the structure of repair junctions manually or with other general-purpose software. Here, we describe methods to produce amplicon libraries of DSB repair junctions for sequencing, to map the sequencing reads, and then to use a robust, custom python script, Hi-FiBR, to analyze the sequence structure of mapped reads. The Hi-FiBR analysis processes large data sets quickly and provides information such as number and type of repair events, size of deletion, size of insertion and inserted sequence, microhomology usage, and whether mismatches are due to sequencing error or biological effect. The analysis also corrects for common alignment errors generated by sequencing read mapping tools, allowing high-throughput analysis of DSB break repair fidelity to be accurately conducted regardless of which suite of NGS analysis software is available.
Keywords: Alternative end joining; Amplicon; DNA double-strand break; Hi-FiBR; High-throughput sequencing; Homologous recombination; Microhomology; Nonhomologous end joining; Read alignment; Rearrangement; Repair junction.
© 2018 Elsevier Inc. All rights reserved.
Figures



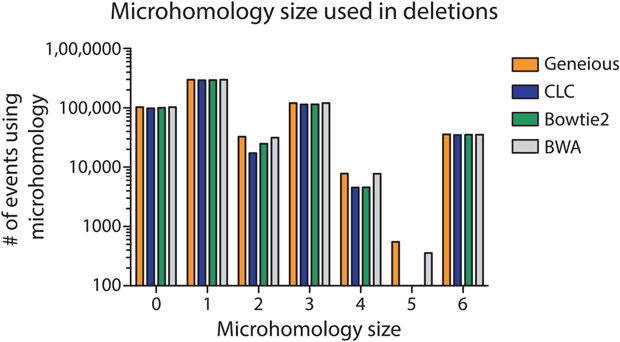
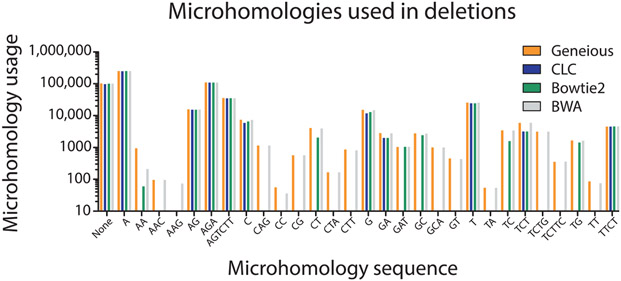
Similar articles
-
Marker-free quantification of repair pathway utilization at Cas9-induced double-strand breaks.Nucleic Acids Res. 2021 May 21;49(9):5095-5105. doi: 10.1093/nar/gkab299. Nucleic Acids Res. 2021. PMID: 33963863 Free PMC article.
-
Development of a novel method to create double-strand break repair fingerprints using next-generation sequencing.DNA Repair (Amst). 2015 Feb;26:44-53. doi: 10.1016/j.dnarep.2014.12.002. Epub 2014 Dec 19. DNA Repair (Amst). 2015. PMID: 25547252
-
Recovery of Alternative End-Joining Repair Products From Drosophila Embryos.Methods Enzymol. 2018;601:91-110. doi: 10.1016/bs.mie.2017.11.027. Epub 2018 Feb 3. Methods Enzymol. 2018. PMID: 29523244
-
Genomic rearrangements induced by unscheduled DNA double strand breaks in somatic mammalian cells.FEBS J. 2017 Aug;284(15):2324-2344. doi: 10.1111/febs.14053. Epub 2017 Mar 22. FEBS J. 2017. PMID: 28244221 Review.
-
Microhomology-mediated end joining: Good, bad and ugly.Mutat Res. 2018 May;809:81-87. doi: 10.1016/j.mrfmmm.2017.07.002. Epub 2017 Jul 16. Mutat Res. 2018. PMID: 28754468 Free PMC article. Review.
Cited by
-
Site-specific targeting of a light activated dCas9-KillerRed fusion protein generates transient, localized regions of oxidative DNA damage.PLoS One. 2020 Dec 17;15(12):e0237759. doi: 10.1371/journal.pone.0237759. eCollection 2020. PLoS One. 2020. PMID: 33332350 Free PMC article.
-
Defining APOBEC-induced mutation signatures and modifying activities in yeast.Methods Enzymol. 2025;713:115-161. doi: 10.1016/bs.mie.2024.11.041. Epub 2025 Apr 2. Methods Enzymol. 2025. PMID: 40250951 Free PMC article.
-
Polymerase δ promotes chromosomal rearrangements and imprecise double-strand break repair.Proc Natl Acad Sci U S A. 2020 Nov 3;117(44):27566-27577. doi: 10.1073/pnas.2014176117. Epub 2020 Oct 19. Proc Natl Acad Sci U S A. 2020. PMID: 33077594 Free PMC article.
-
Parp3 promotes long-range end joining in murine cells.Proc Natl Acad Sci U S A. 2018 Oct 2;115(40):10076-10081. doi: 10.1073/pnas.1801591115. Epub 2018 Sep 13. Proc Natl Acad Sci U S A. 2018. PMID: 30213852 Free PMC article.
-
Marker-free quantification of repair pathway utilization at Cas9-induced double-strand breaks.Nucleic Acids Res. 2021 May 21;49(9):5095-5105. doi: 10.1093/nar/gkab299. Nucleic Acids Res. 2021. PMID: 33963863 Free PMC article.
References
-
- Beagan K, Armstrong RL, Witsell A, Roy U, Renedo N, Baker AE, et al. (2017). Drosophila DNA polymerase theta utilizes both helicase-like and polymerase domains during microhomology-mediated end joining and interstrand crosslink repair. PLoS Genetics, 13(5), e1006813. 10.1371/journal.pgen.1006813. - DOI - PMC - PubMed
-
- Bennett CB, Lewis AL, Baldwin KK, & Resnick MA (1993). Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proceedings of the National Academy of Sciences of the United States of America, 90(12), 5613–5617. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8516308. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
