

Integrating Experimental and In Silico HTS in the Discovery of Inhibitors of Protein-Nucleic Acid Interactions
- PMID: 29523234
- PMCID: PMC6010167
- DOI: 10.1016/bs.mie.2017.11.036
Integrating Experimental and In Silico HTS in the Discovery of Inhibitors of Protein-Nucleic Acid Interactions
Abstract
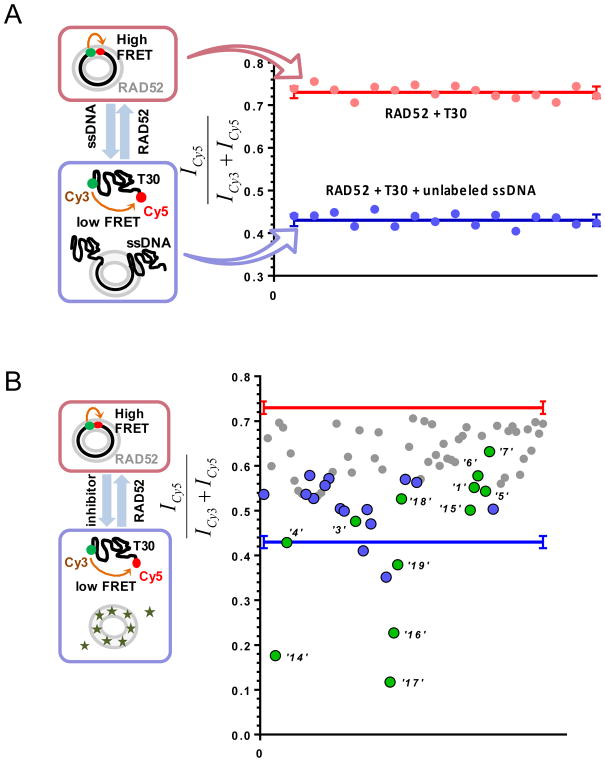
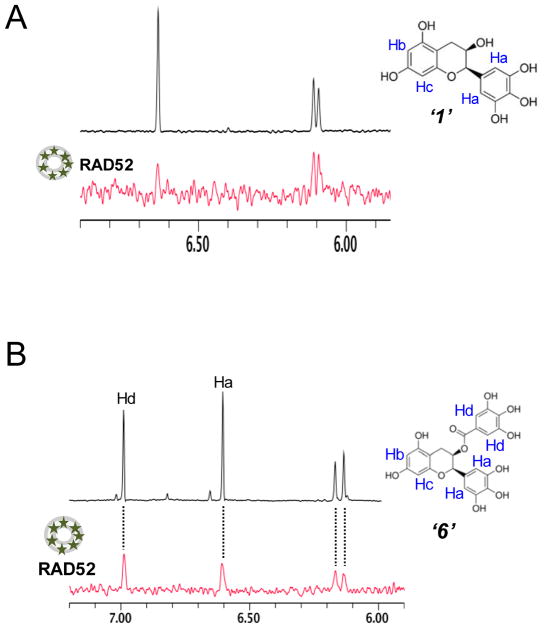
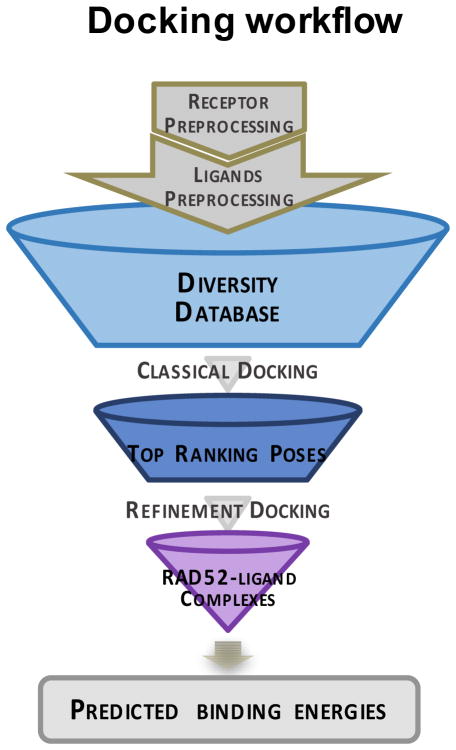
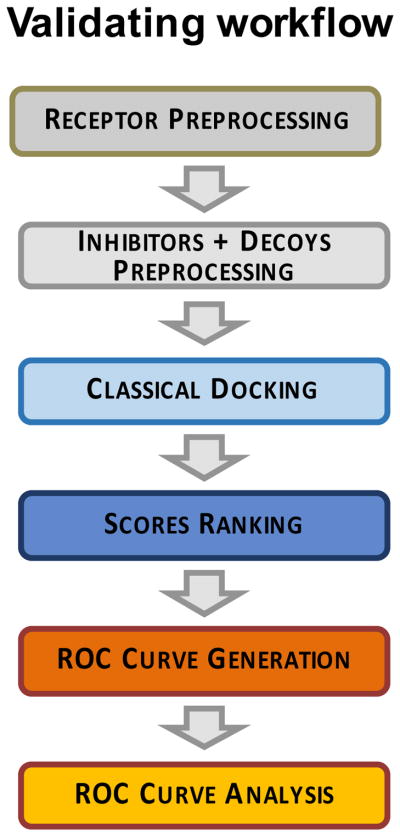
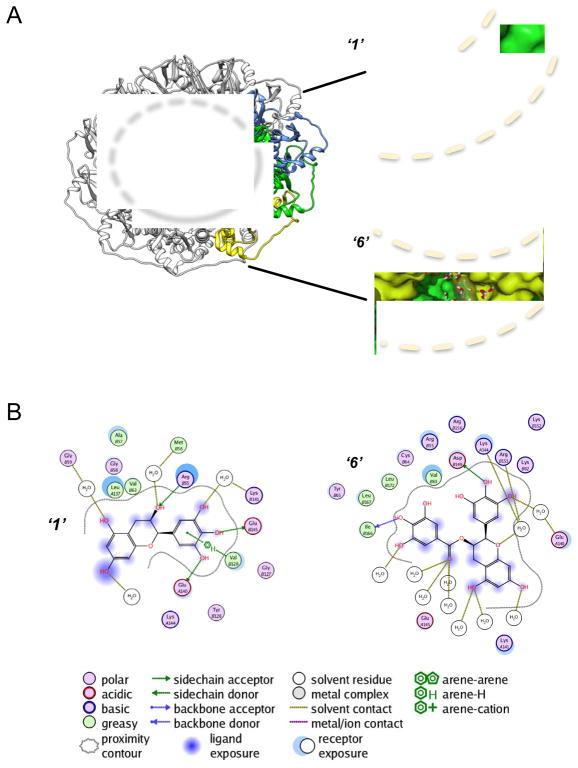
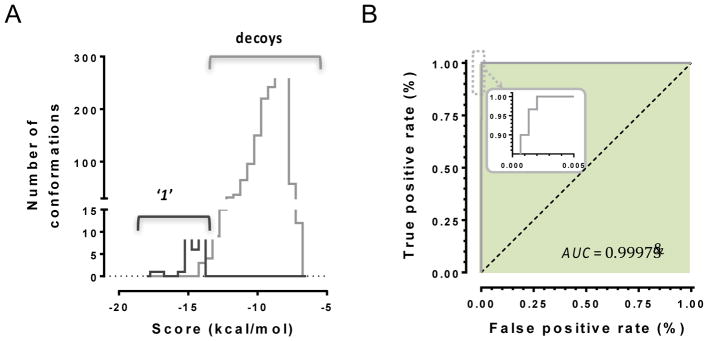
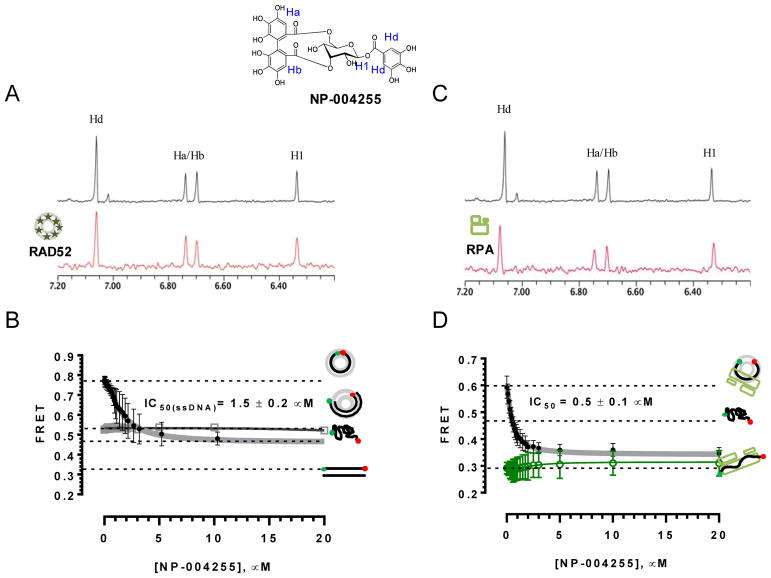
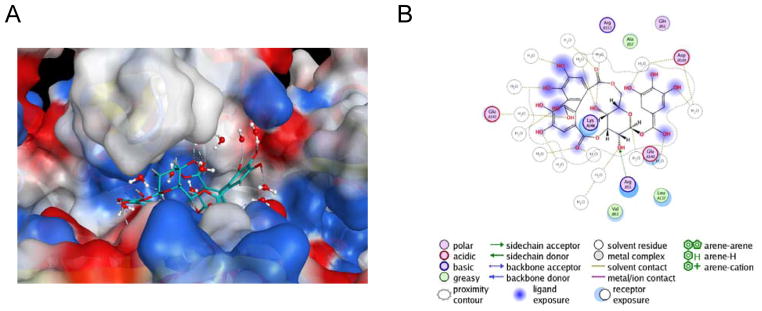
Discovery of novel tool compounds and drug leads against a range of unorthodox protein targets has pushed both experimental screening methodologies as well as the field of structure-based design to the limit in recent years. Increasingly, it has been recognized that some of the most desirable targets for the development of small-molecule effectors are actually protein-protein and protein-nucleic acid interactions. There are numerous nontrivial challenges to pursuing small-molecule lead compounds directed toward PPIs and PNIs: relatively shallow cavities, large surface areas that are natively complexed to macromolecules, complex patterns of interstitial waters, a paucity of "hot spots," large conformational changes upon ligand binding, etc. Although there have been some notable successes targeting PPIs in the last decade, there has been distinctly less success in the realm of targeting PNIs. This chapter focuses on an approach, successfully applied by our group to address the challenge of gaining traction on the PPI target RAD52, which is a protein that binds both single-stranded and double-stranded DNA, and is an anticancer target for certain types of cancer. There are many approaches to tackling the difficult problems of finding effective small molecules that disrupt PPIs and PNIs, but the methods presented here offer a series of elegant solutions, which integrate experimental HTS, biophysical methods, docking, and molecular dynamics in a powerful way. Additionally, the structural knowledge gained from these studies provides a means for rationally understanding what features lead to ligand affinity in these fascinating and highly unorthodox pockets.
Keywords: In silico screening; Natural products; Protein–nucleic acid interactions; Structure-based drug discovery; Virtual screening.
© 2018 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Benchmark Study Based on 2P2IDB to Gain Insights into the Discovery of Small-Molecule PPI Inhibitors.J Phys Chem B. 2018 Mar 8;122(9):2544-2555. doi: 10.1021/acs.jpcb.7b12658. Epub 2018 Feb 22. J Phys Chem B. 2018. PMID: 29420886
-
Natural products used as a chemical library for protein-protein interaction targeted drug discovery.J Mol Graph Model. 2018 Jan;79:46-58. doi: 10.1016/j.jmgm.2017.10.015. Epub 2017 Oct 27. J Mol Graph Model. 2018. PMID: 29136547
-
Protein-protein interaction inhibitors: advances in anticancer drug design.Expert Opin Drug Discov. 2016 Oct;11(10):957-68. doi: 10.1080/17460441.2016.1223038. Epub 2016 Sep 2. Expert Opin Drug Discov. 2016. PMID: 27554357 Review.
-
Computational Screening and Design for Compounds that Disrupt Protein-protein Interactions.Curr Top Med Chem. 2017;17(23):2703-2714. doi: 10.2174/1568026617666170508153904. Curr Top Med Chem. 2017. PMID: 28482793 Review.
-
Virtual screening of small molecules databases for discovery of novel PARP-1 inhibitors: combination of in silico and in vitro studies.J Biomol Struct Dyn. 2017 Jul;35(9):1899-1915. doi: 10.1080/07391102.2016.1199328. Epub 2016 Jul 17. J Biomol Struct Dyn. 2017. PMID: 27315035
Cited by
-
Therapeutic disruption of RAD52-ssDNA complexation via novel drug-like inhibitors.NAR Cancer. 2023 May 1;5(2):zcad018. doi: 10.1093/narcan/zcad018. eCollection 2023 Jun. NAR Cancer. 2023. PMID: 37139244 Free PMC article.
-
Decrypting a cryptic allosteric pocket in H. pylori glutamate racemase.Commun Chem. 2021 Dec 10;4(1):172. doi: 10.1038/s42004-021-00605-z. Commun Chem. 2021. PMID: 36697800
-
POT1 stability and binding measured by fluorescence thermal shift assays.PLoS One. 2021 Mar 30;16(3):e0245675. doi: 10.1371/journal.pone.0245675. eCollection 2021. PLoS One. 2021. PMID: 33784306 Free PMC article.
-
Decrypting a Cryptic Allosteric Pocket in H. pylori Glutamate Racemase.Commun Chem. 2021;4:172. doi: 10.1038/s42004-021-00605-z. Epub 2021 Dec 10. Commun Chem. 2021. PMID: 35673630 Free PMC article.
References
-
- Chemical Computing Group Inc. Molecular Operating Environment. 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2013.08 ed.
-
- ALEXANDRE VARNEK AT. Chemoinformatics Approaches to Virtual Screening. Royal Society of Chemistry; 2008.
-
- DE VEGA MJ, MARTIN-MARTINEZ M, GONZALEZ-MUNIZ R. Modulation of protein-protein interactions by stabilizing/mimicking protein secondary structure elements. Curr Top Med Chem. 2007;7:33–62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
