

Loss of FFAR2 promotes colon cancer by epigenetic dysregulation of inflammation suppressors
- PMID: 29524208
- PMCID: PMC6041131
- DOI: 10.1002/ijc.31366
Loss of FFAR2 promotes colon cancer by epigenetic dysregulation of inflammation suppressors
Abstract
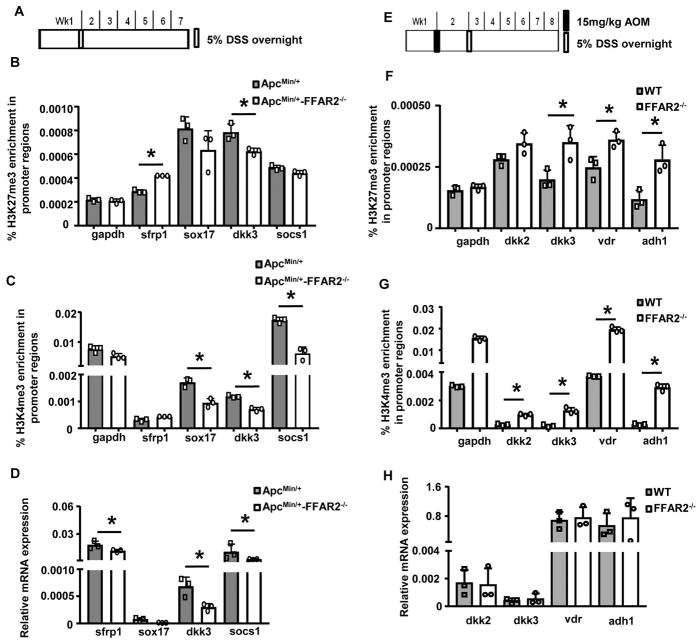
Free fatty acid receptor 2 (FFAR2, also named GPR43), is activated by short-chain fatty acids (SCFAs), such as butyrate, that are produced when gut bacteria ferment dietary fiber. FFAR2 has been suggested to regulate colonic inflammation, which is a major risk factor for the development of colon cancer and is also linked to epigenetic dysregulation in colon carcinogenesis. The current study assessed whether FFAR2, acting as an epigenetic regulator, protects against colon carcinogenesis. To mimic the mild inflammation that promotes human colon cancer, we treated mice with dextran sodium sulfate (DSS) overnight, which avoids excessive inflammation but induces mild inflammation that promotes colon carcinogenesis in the ApcMin/+ and the azoxymethane (AOM)-treated mice. Our results showed that FFAR2 deficiency promotes the development of colon adenoma in the ApcMin/+ /DSS mice and the progression of adenoma to adenocarcinoma in the AOM/DSS mice. FFAR2's downstream cAMP-PKA-CREB pathway was enhanced, leading to overexpression of histone deacetylases (HDACs) in the FFAR2-deficient mice. ChIP-qPCR analysis revealed differential binding of H3K27me3 and H3K4me3 histone marks onto the promoter regions of inflammation suppressors (e.g., sfrp1, dkk3, socs1), resulting in decreased expression of these genes in the FFAR2-deficient mice. Also, more neutrophils infiltrated into tumors and colon lamina propria of the FFAR2-deficient mice. Depletion of neutrophils blocked the progression of colon tumors. In addition, FFAR2 is required for butyrate to suppress HDAC expression and hypermethylation of inflammation suppressors. Therefore, our results suggest that FFAR2 is an epigenetic tumor suppressor that acts at multiple stages of colon carcinogenesis.
Keywords: FFAR2; HDAC; butyrate; colon cancer; epigenetics.
© 2018 UICC.
Conflict of interest statement
No potential conflict of interest was disclosed.
Figures




References
-
- Lee T, Schwandner R, Swaminath G, Weiszmann J, Cardozo M, Greenberg J, Jaeckel P, Ge H, Wang Y, Jiao X, Liu J, Kayser F, et al. Identification and functional characterization of allosteric agonists for the G protein-coupled receptor FFA2. Mol Pharmacol. 2008;74:1599–609. - PubMed
-
- Shao RH, Tian X, Gorgun G, Urbano AG, Foss FM. Arginine butyrate increases the cytotoxicity of DAB389IL-2 in leukemia and lymphoma cells by upregulation of IL-2Rβ gene. Leuk Res. 2002;26:1077–83. - PubMed
-
- Lakshmaiah KC, Jacob LA, Aparna S, Lokanatha D, Saldanha SC. Epigenetic therapy of cancer with histone deacetylase inhibitors. J Cancer Res Ther. 2014;10:469–78. - PubMed
-
- Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. 2014;121:91–119. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
