

Hyperhomocysteinemia potentiates diabetes-impaired EDHF-induced vascular relaxation: Role of insufficient hydrogen sulfide
- PMID: 29524844
- PMCID: PMC5854893
- DOI: 10.1016/j.redox.2018.02.006
Hyperhomocysteinemia potentiates diabetes-impaired EDHF-induced vascular relaxation: Role of insufficient hydrogen sulfide
Abstract
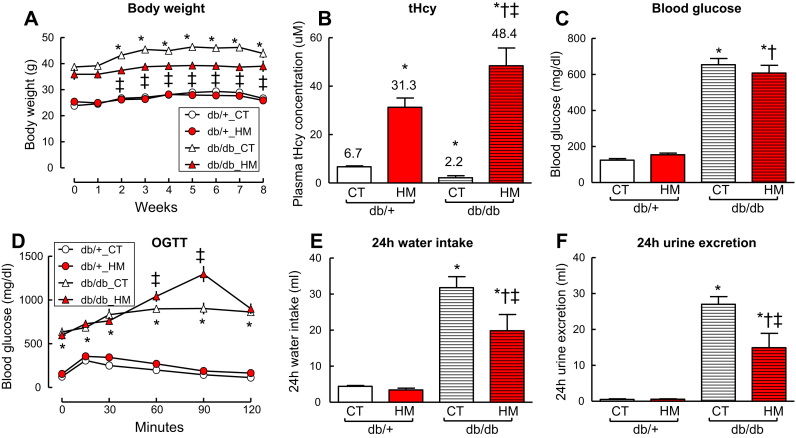
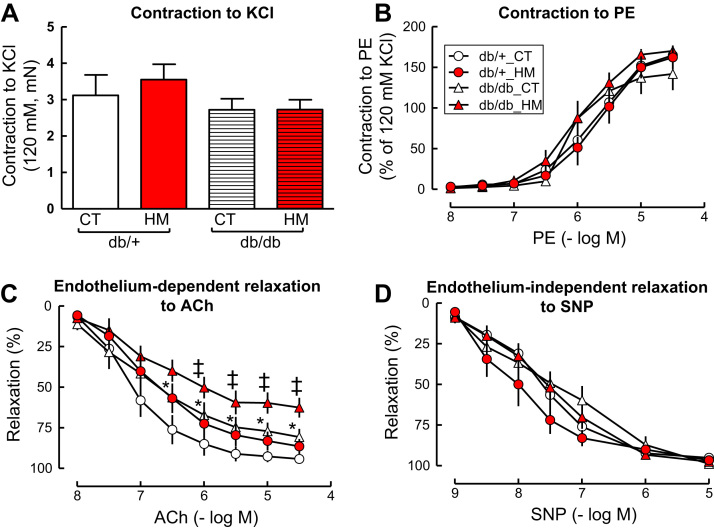
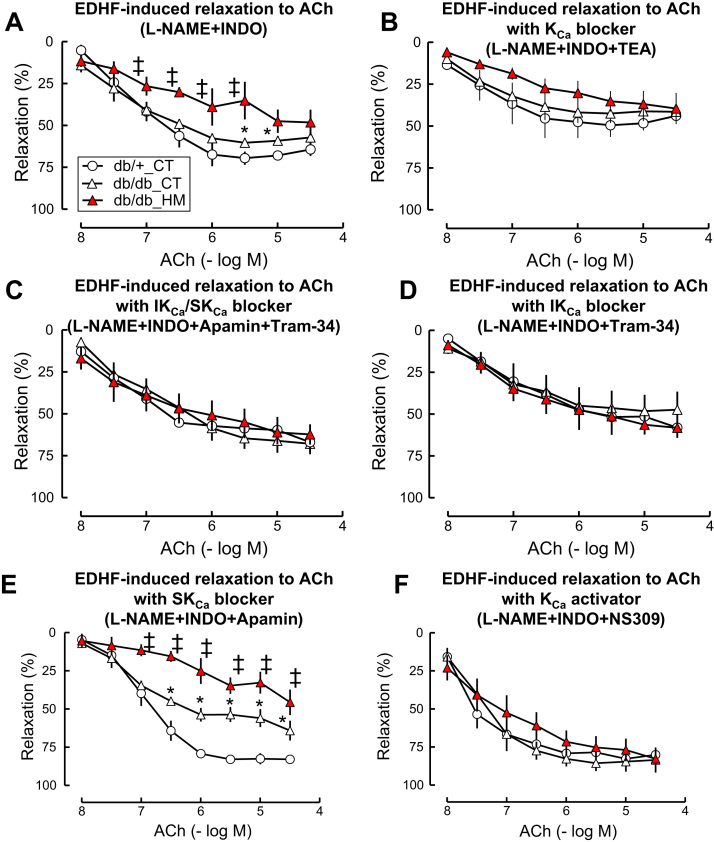
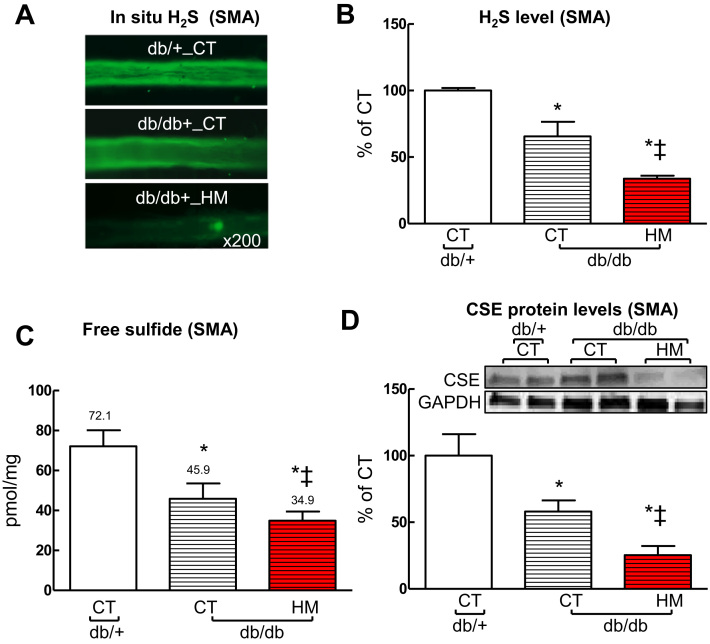
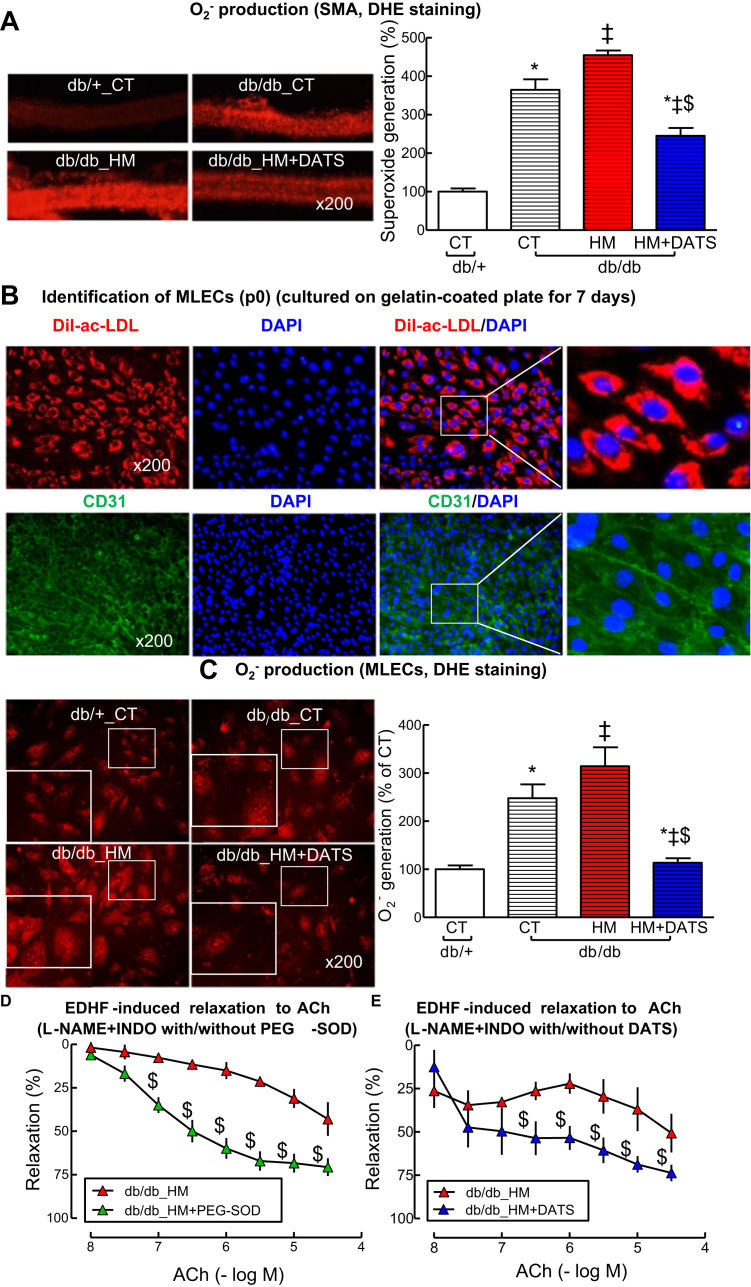
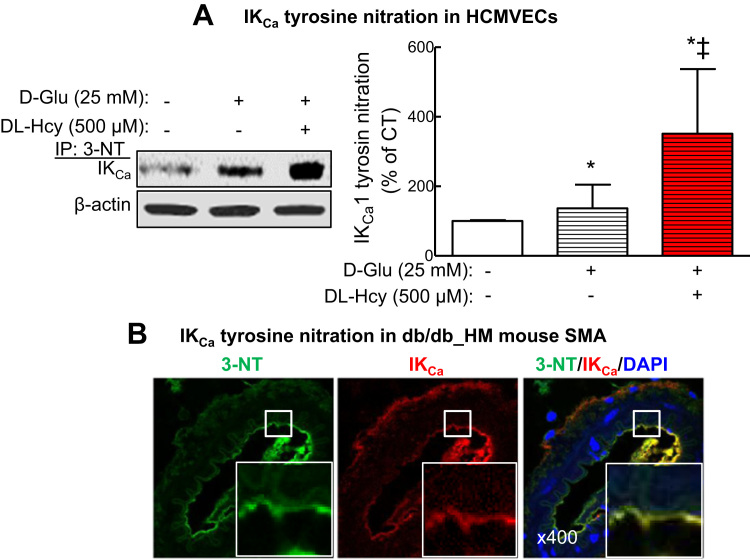
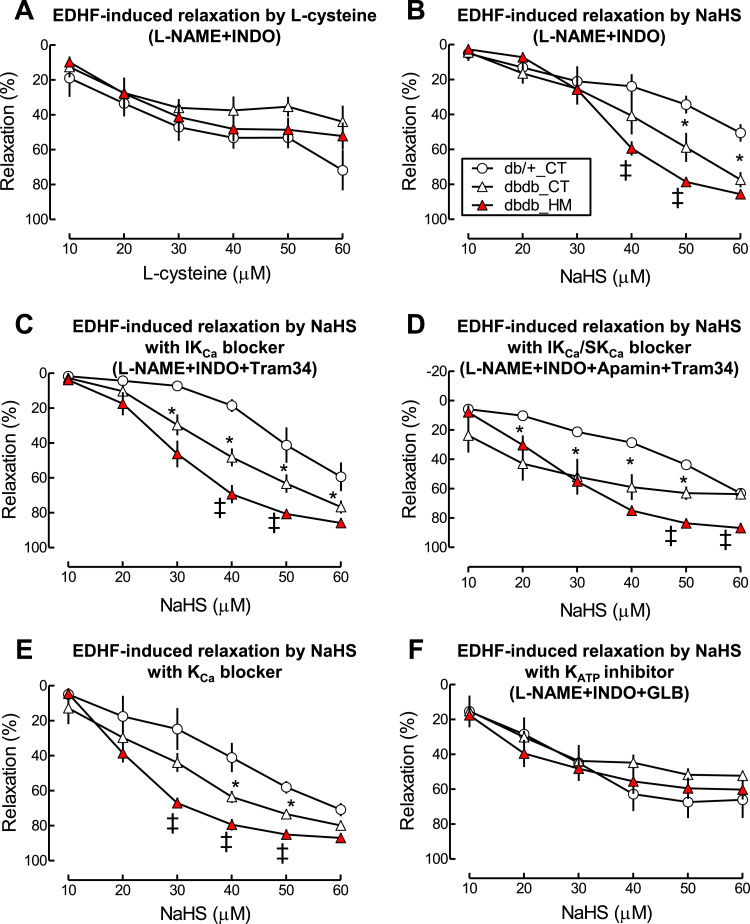
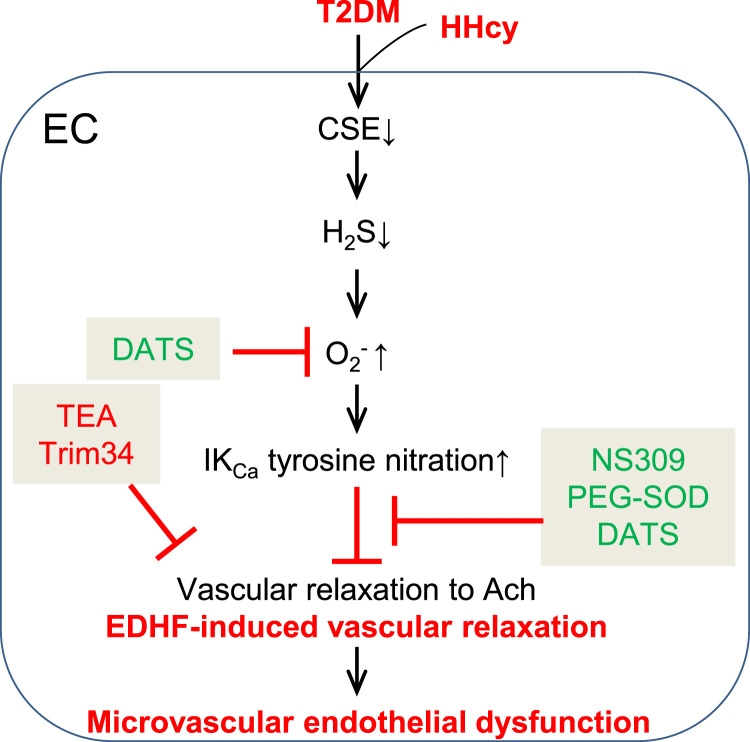
Insufficient hydrogen sulfide (H2S) has been implicated in Type 2 diabetic mellitus (T2DM) and hyperhomocysteinemia (HHcy)-related cardiovascular complications. We investigated the role of H2S in T2DM and HHcy-induced endothelial dysfunction in small mesenteric artery (SMA) of db/db mice fed a high methionine (HM) diet. HM diet (8 weeks) induced HHcy in both T2DM db/db mice and non-diabetic db/+ mice (total plasma Hcy: 48.4 and 31.3 µM, respectively), and aggravated the impaired endothelium-derived hyperpolarization factor (EDHF)-induced endothelium-dependent relaxation to acetylcholine (ACh), determined by the presence of eNOS inhibitor N(ω)-nitro-L-arginine methyl ester (L-NAME) and prostacyclin (PGI2) inhibitor indomethacin (INDO), in SMA from db/db mice but not that from db/+ mice. A non-selective Ca2+-active potassium channel (KCa) opener NS309 rescued T2DM/HHcy-impaired EDHF-mediated vascular relaxation to ACh. EDHF-induced relaxation to ACh was inhibited by a non-selective KCa blocker TEA and intermediate-conductance KCa blocker (IKCa) Tram-34, but not by small-conductance KCa (SKCa) blocker Apamin. HHcy potentiated the reduction of free sulfide, H2S and cystathionine γ-lyase protein, which converts L-cysteine to H2S, in SMA of db/db mice. Importantly, a stable H2S donor DATS diminished the enhanced O2- production in SMAs and lung endothelial cells of T2DM/HHcy mice. Antioxidant PEG-SOD and DATS improved T2DM/HHcy impaired relaxation to ACh. Moreover, HHcy increased hyperglycemia-induced IKCa tyrosine nitration in human micro-vascular endothelial cells. EDHF-induced vascular relaxation to L-cysteine was not altered, whereas such relaxation to NaHS was potentiated by HHcy in SMA of db/db mice which was abolished by ATP-sensitive potassium channel blocker Glycolamide but not by KCa blockers.
Conclusions: Intermediate HHcy potentiated H2S reduction via CSE-downregulation in microvasculature of T2DM mice. H2S is justified as an EDHF. Insufficient H2S impaired EDHF-induced vascular relaxation via oxidative stress and IKCa inactivation in T2DM/HHcy mice. H2S therapy may be beneficial for prevention and treatment of micro-vascular complications in patients with T2DM and HHcy.
Keywords: Calcium-activated potassium channel (K(Ca)); Endothelial dysfunction; Hydrogen sulfide; Micro-vasculature; T2DM.
Copyright © 2018 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Gregg E.W., Li Y., Wang J., Burrows N.R., Ali M.K., Rolka D., Williams D.E., Geiss L. Changes in diabetes-related complications in the United States, 1990–2010. N. Engl. J. Med. 2014;370:1514–1523. - PubMed
-
- Unwin N., Gan D., Whiting D. The IDF Diabetes Atlas: providing evidence, raising awareness and promoting action. Diabetes Res. Clin. Pract. 2010;87:2–3. - PubMed
-
- Furchgott R.F., Zawadzki J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. - PubMed
-
- Feletou M., Vanhoutte P.M. Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler. Thromb. Vasc. Biol. 2006;26:1215–1225. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL126186/HL/NHLBI NIH HHS/United States
- R01 HL077288/HL/NHLBI NIH HHS/United States
- R01 HL130233/HL/NHLBI NIH HHS/United States
- R01 HL126952/HL/NHLBI NIH HHS/United States
- R01 HL131460/HL/NHLBI NIH HHS/United States
- R01 HL082774/HL/NHLBI NIH HHS/United States
- R01 HL110764/HL/NHLBI NIH HHS/United States
- R01 DK104116/DK/NIDDK NIH HHS/United States
- R37 HL053354/HL/NHLBI NIH HHS/United States
- R01 HL132399/HL/NHLBI NIH HHS/United States
- R01 HL091983/HL/NHLBI NIH HHS/United States
- R01 HL138749/HL/NHLBI NIH HHS/United States
- P01 HL108795/HL/NHLBI NIH HHS/United States
- R01 DK113775/DK/NIDDK NIH HHS/United States
- R01 HL117654/HL/NHLBI NIH HHS/United States
- P01 HL134608/HL/NHLBI NIH HHS/United States
- R01 HL067033/HL/NHLBI NIH HHS/United States
- R01 HL053354/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
