

Insulin resistance causes inflammation in adipose tissue
- PMID: 29528335
- PMCID: PMC5873875
- DOI: 10.1172/JCI96139
Insulin resistance causes inflammation in adipose tissue
Abstract
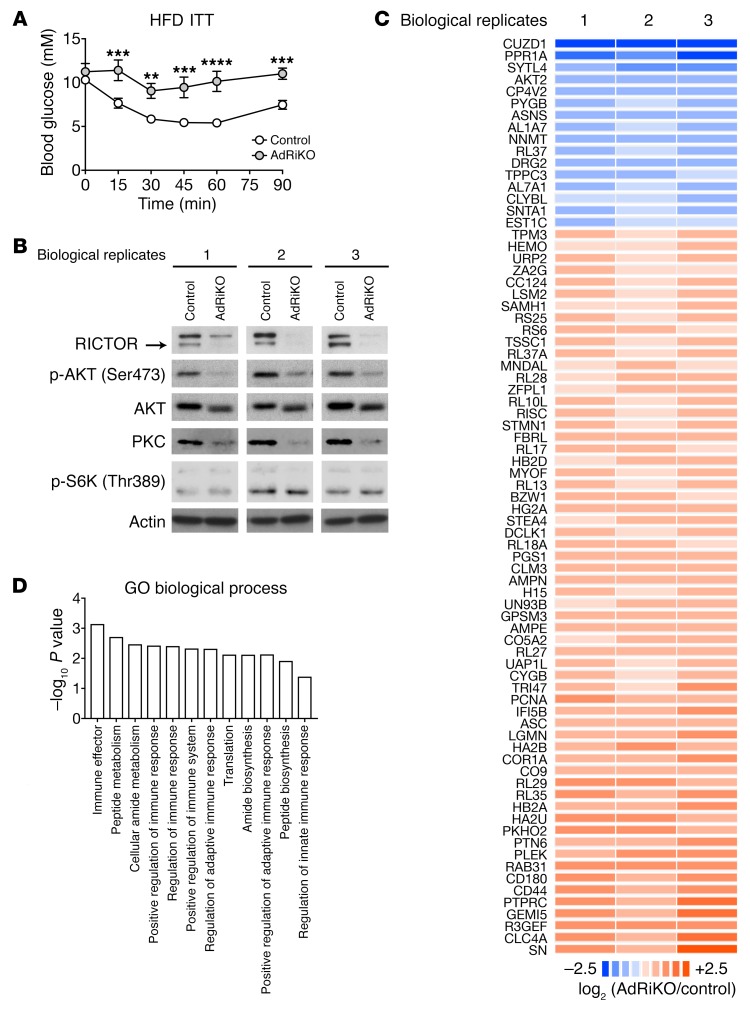
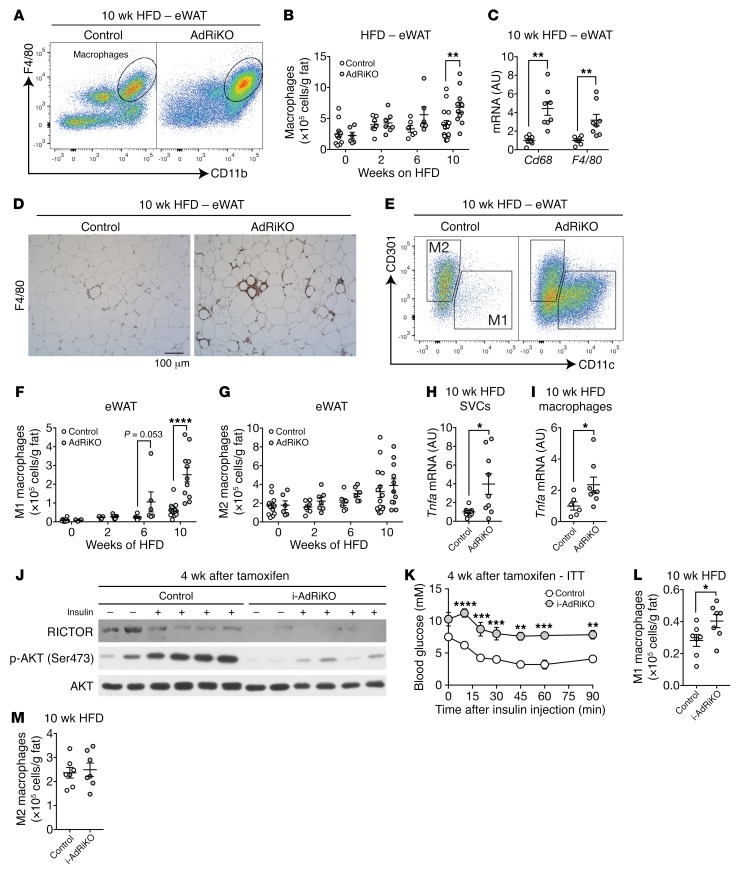
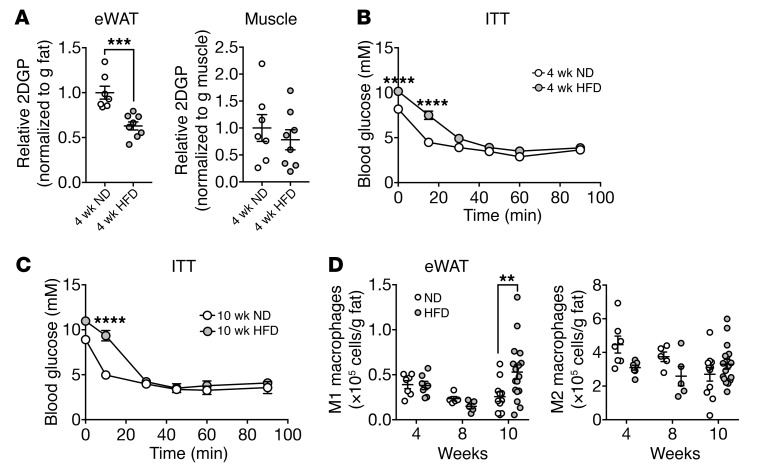
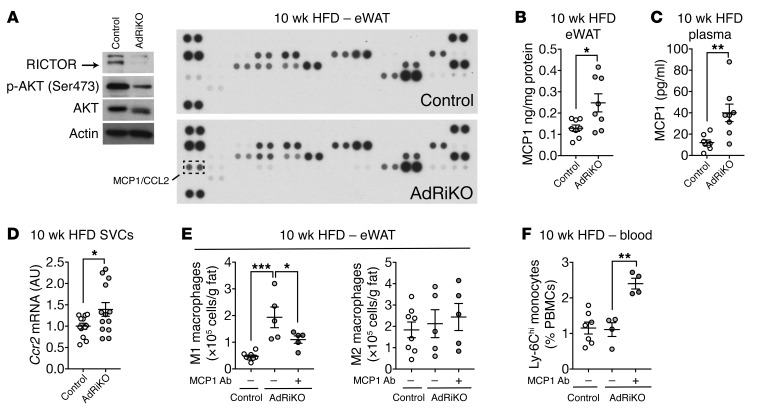
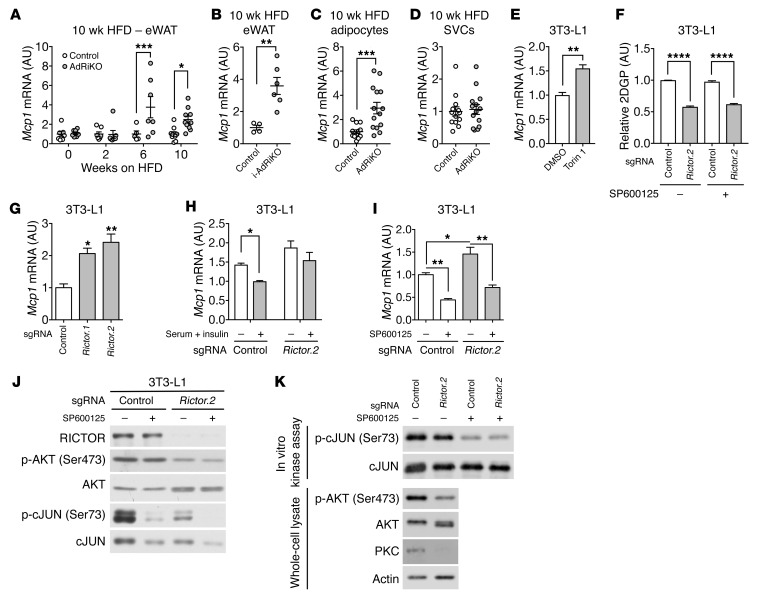
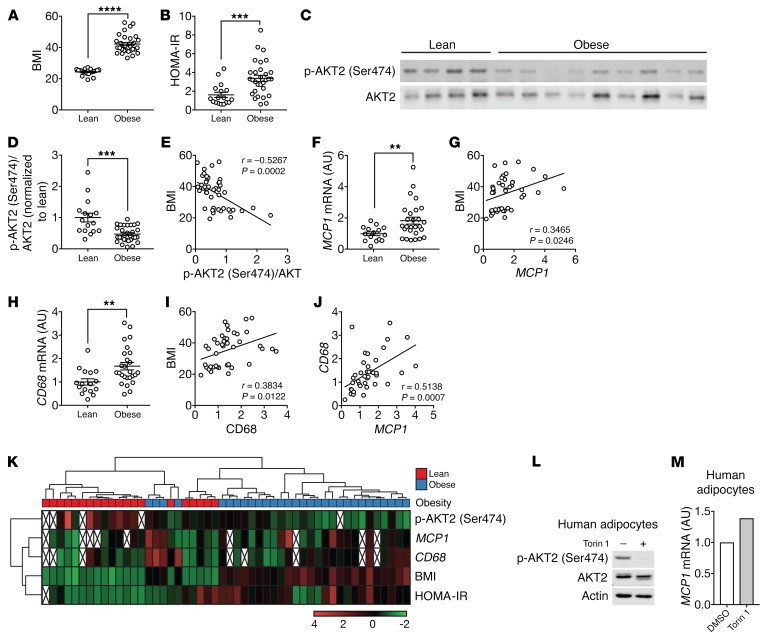
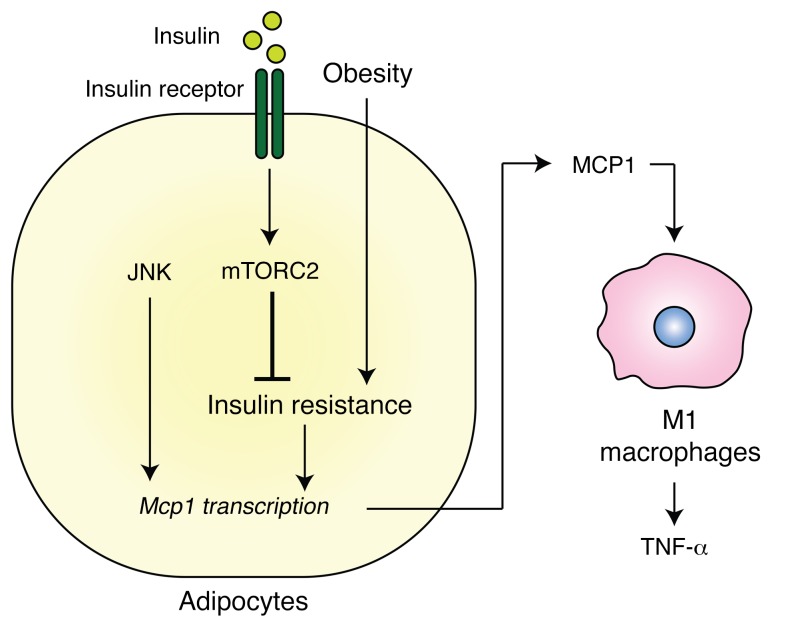
Obesity is a major risk factor for insulin resistance and type 2 diabetes. In adipose tissue, obesity-mediated insulin resistance correlates with the accumulation of proinflammatory macrophages and inflammation. However, the causal relationship of these events is unclear. Here, we report that obesity-induced insulin resistance in mice precedes macrophage accumulation and inflammation in adipose tissue. Using a mouse model that combines genetically induced, adipose-specific insulin resistance (mTORC2-knockout) and diet-induced obesity, we found that insulin resistance causes local accumulation of proinflammatory macrophages. Mechanistically, insulin resistance in adipocytes results in production of the chemokine monocyte chemoattractant protein 1 (MCP1), which recruits monocytes and activates proinflammatory macrophages. Finally, insulin resistance (high homeostatic model assessment of insulin resistance [HOMA-IR]) correlated with reduced insulin/mTORC2 signaling and elevated MCP1 production in visceral adipose tissue from obese human subjects. Our findings suggest that insulin resistance in adipose tissue leads to inflammation rather than vice versa.
Keywords: Adipose tissue; Inflammation; Macrophages; Metabolism; Obesity.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
