

ChIP-ping the branches of the tree: functional genomics and the evolution of eukaryotic gene regulation
- PMID: 29529131
- PMCID: PMC5889016
- DOI: 10.1093/bfgp/ely004
ChIP-ping the branches of the tree: functional genomics and the evolution of eukaryotic gene regulation
Abstract
Advances in the methods for detecting protein-DNA interactions have played a key role in determining the directions of research into the mechanisms of transcriptional regulation. The most recent major technological transformation happened a decade ago, with the move from using tiling arrays [chromatin immunoprecipitation (ChIP)-on-Chip] to high-throughput sequencing (ChIP-seq) as a readout for ChIP assays. In addition to the numerous other ways in which it is superior to arrays, by eliminating the need to design and manufacture them, sequencing also opened the door to carrying out comparative analyses of genome-wide transcription factor occupancy across species and studying chromatin biology in previously less accessible model and nonmodel organisms, thus allowing us to understand the evolution and diversity of regulatory mechanisms in unprecedented detail. Here, we review the biological insights obtained from such studies in recent years and discuss anticipated future developments in the field.
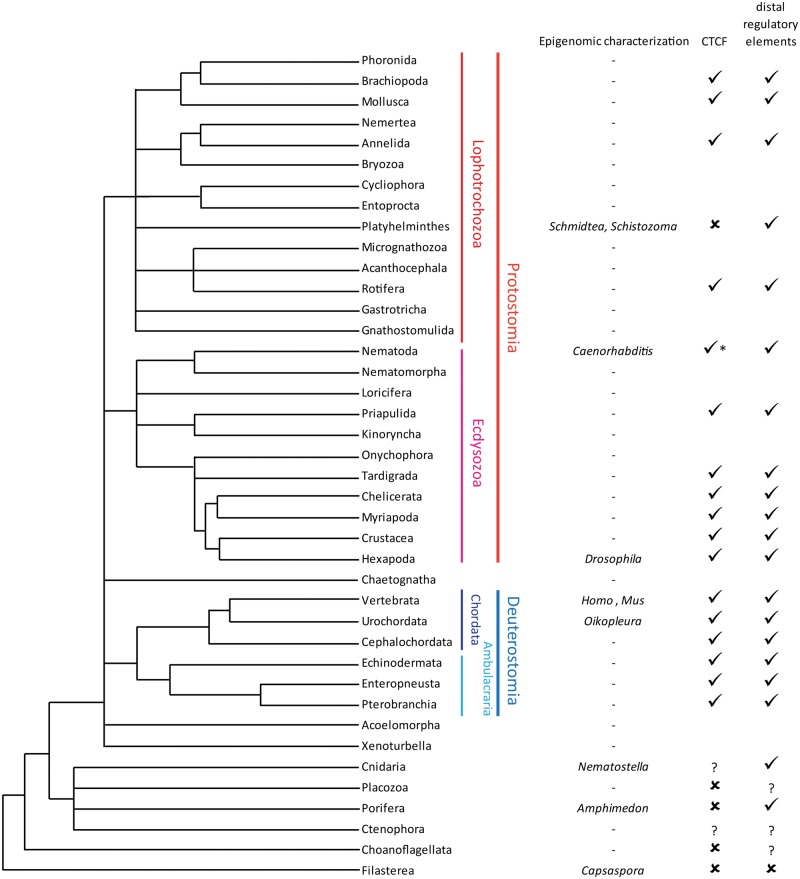
Figures
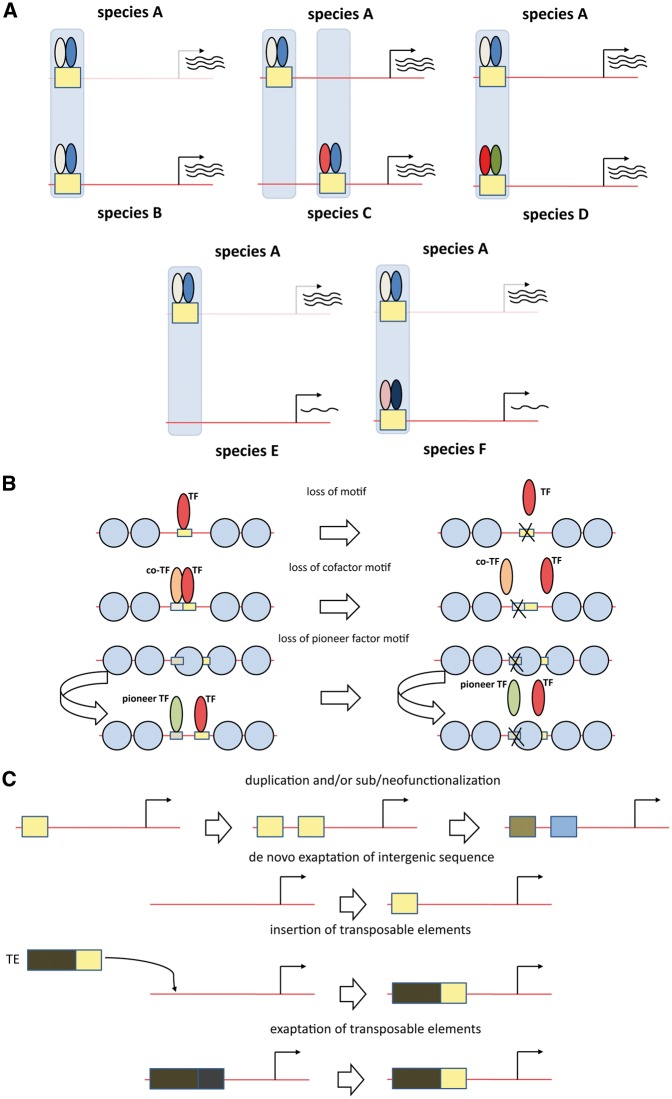
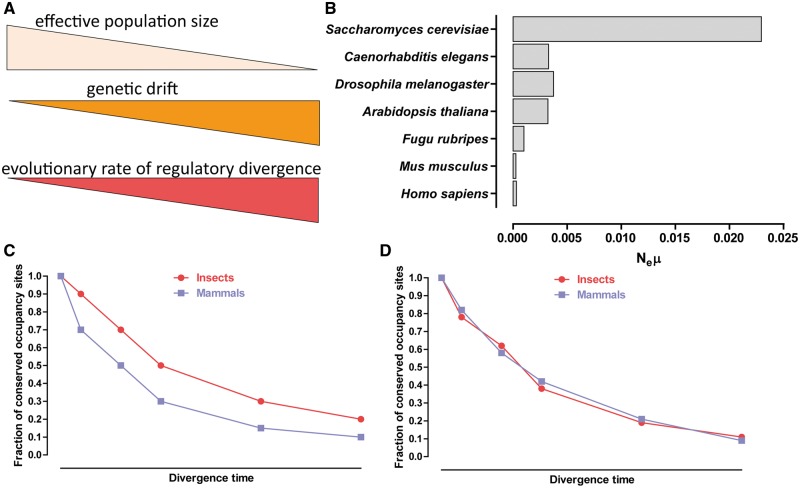
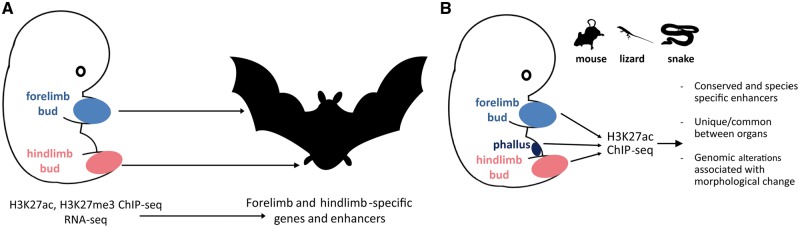
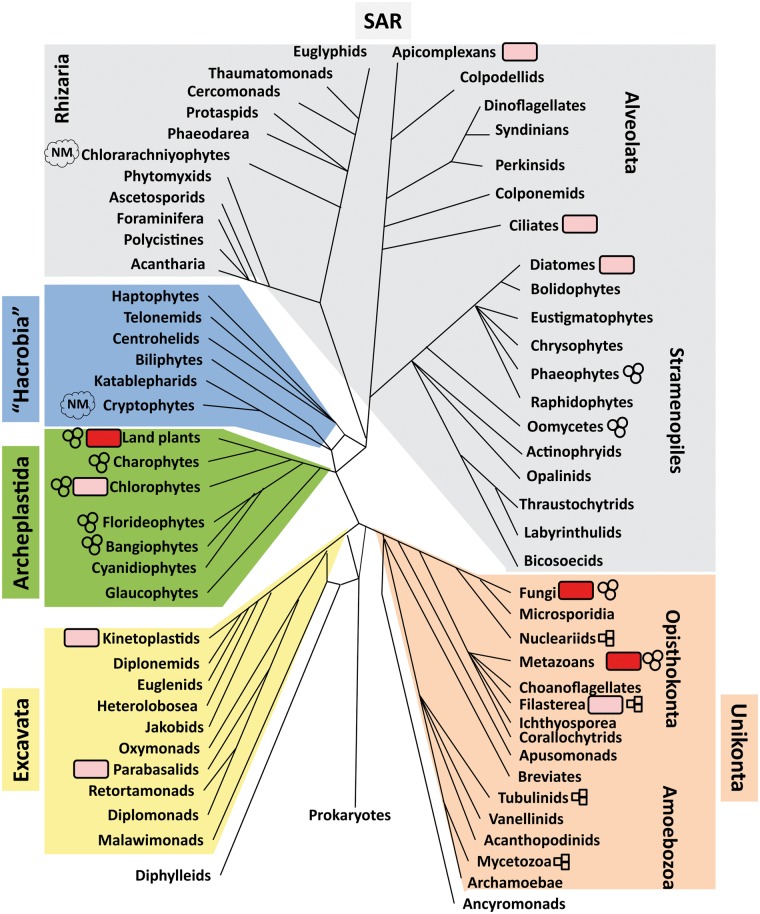
References
-
- Solomon MJ, Larsen PL, Varshavsky A.. Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell 1988;53(6):937–47. - PubMed
-
- Hecht A, Strahl-Bolsinger S, Grunstein M.. Spreading of transcriptional repressor SIR3 from telomeric heterochromatin. Nature 1996;383(6595):92–6. - PubMed
-
- Ren B, Robert F, Wyrick JJ, et al.Genome-wide location and function of DNA binding proteins. Science 2000;290(5500):2306–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
