

Comment
doi: 10.1093/brain/awy024.
The expanding neurological phenotype of DNM1L-related disorders
Affiliations
- PMID: 29529134
- PMCID: PMC11505533
- DOI: 10.1093/brain/awy024
Item in Clipboard
Comment
The expanding neurological phenotype of DNM1L-related disorders
Brain.
.
No abstract available
Figures
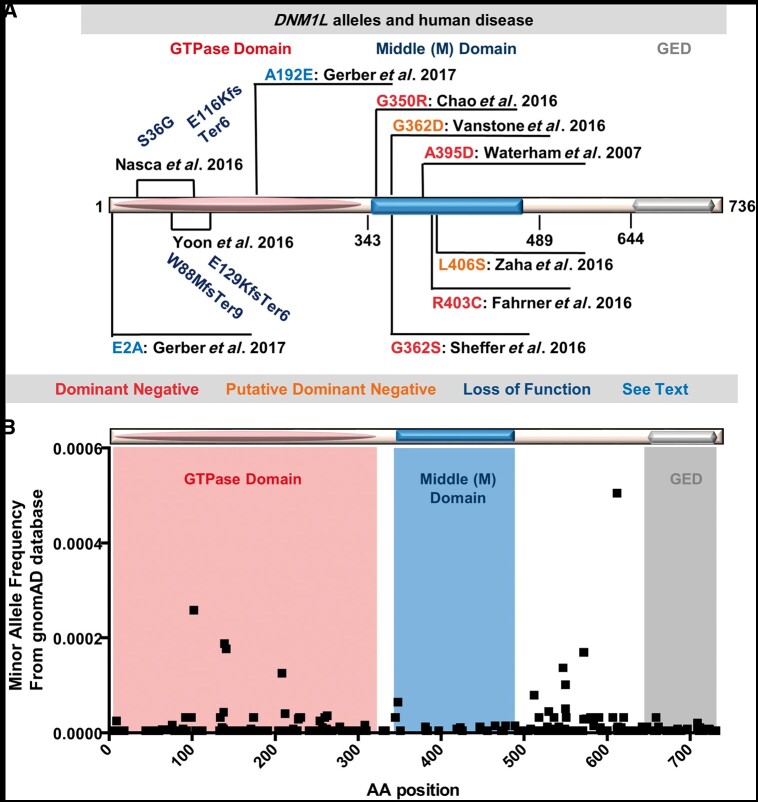
DNM1L alleles in humans. (A) DNM1L alleles associated with human disease and their effects on gene function. Red: dominant negative heterozygous mutations associated with early onset disease such as encephalopathy due to defective mitochondrial and peroxisomal fission-1 (EMPF1) and experimentally validated in cells or animal models as dominant negative alleles. Orange: putative dominant negative heterozygous mutations associated with early disease. Dark blue: Compound heterozygous mutations resulting in loss of function in cases with bi-allelic variants in DNM1L. Light blue: heterozygous mutations associated with isolated optic atrophy reported by Gerber et al. (B) A graph of minor allele frequency on the y-axis versus the encoded amino acid position in the DNM1L protein on the x-axis for all the missense allele calls available in the gnomAD (gnomad.broadinstitute.org/) a database of >120 000 individuals. An approximate overlay of the domains encoded by the amino acids for the GTPase, middle domain and GED (effector domain) are shown in salmon, blue and grey, respectively.
Comment in
-
Reply: The expanding neurological phenotype of DNM1L-related disorders.Brain. 2018 Apr 1;141(4):e29. doi: 10.1093/brain/awy027. Brain. 2018. PMID: 29529130 No abstract available.
Comment on
-
Mutations in DNM1L, as in OPA1, result in dominant optic atrophy despite opposite effects on mitochondrial fusion and fission.Brain. 2017 Oct 1;140(10):2586-2596. doi: 10.1093/brain/awx219. Brain. 2017. PMID: 28969390
References
-
- Gerber S, Charif M, Chevrollier A, Chaumette T, Angebault C, Kane MS, et al. Mutations in DNM1L, as in OPA1, result indominant optic atrophy despite opposite effectson mitochondrial fusion and fission. Brain 2017; 140: 2586–96. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
