

Hepatitis A Virus Codon Usage: Implications for Translation Kinetics and Capsid Folding
- PMID: 29530949
- PMCID: PMC6169985
- DOI: 10.1101/cshperspect.a031781
Hepatitis A Virus Codon Usage: Implications for Translation Kinetics and Capsid Folding
Abstract
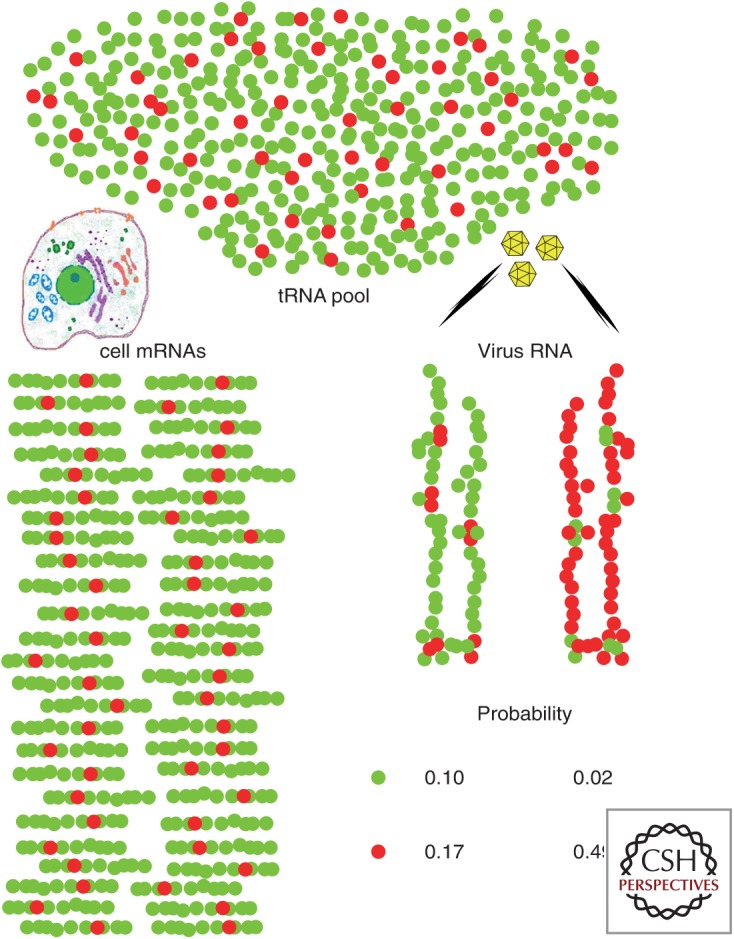
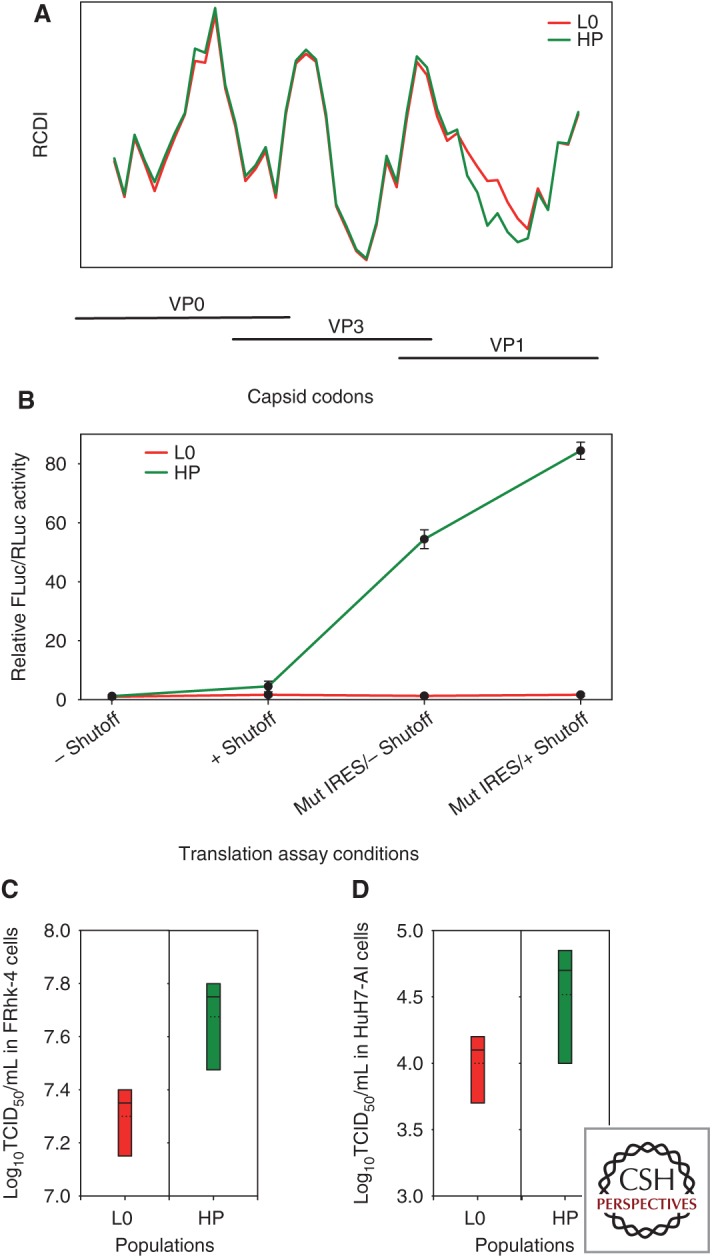
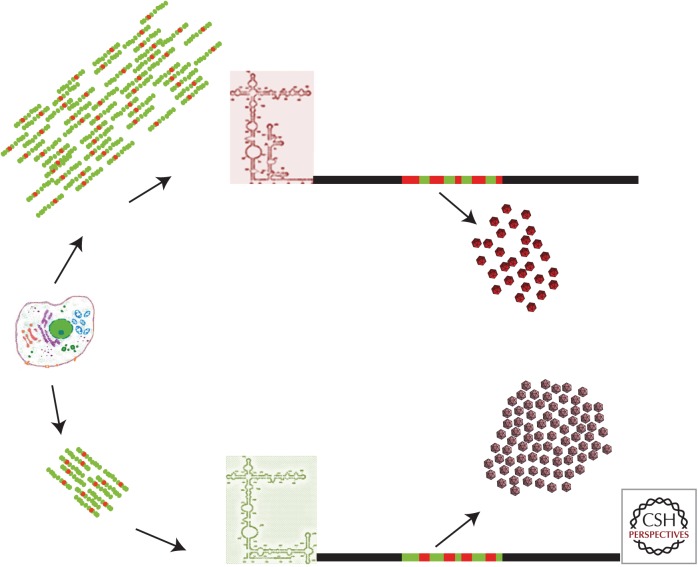
Codon usage bias is universal to all genomes. Hepatitis A virus (HAV) codon usage is highly biased and deoptimized with respect to its host. Accordingly, HAV is unable to induce cellular translational shutoff and its internal ribosome entry site (IRES) is inefficient. Codon usage deoptimization may be seen as a hawk (host cell) versus dove (HAV) game strategy for accessing transfer RNA (tRNA). HAV avoids use of abundant host cell codons and thereby eludes competition for the corresponding tRNAs. Instead, codons that are abundant or rare in cellular messenger RNAs (mRNAs) are used relatively rarely in its genome, although intermediately abundant host cell codons are abundant in the viral genome. Rare codons in the capsid coding region slow down the translation elongation rate, and in doing so intrinsically modulate capsid folding, which is critical to the stability of a virus transmitted through the fecal-oral route. HAV is a paradigmatic example of what has been proposed as a codon usage "code" for protein structure.
Copyright © 2018 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources