

Classification and Genomic Diversity of Enterically Transmitted Hepatitis Viruses
- PMID: 29530950
- PMCID: PMC6120691
- DOI: 10.1101/cshperspect.a031880
Classification and Genomic Diversity of Enterically Transmitted Hepatitis Viruses
Abstract
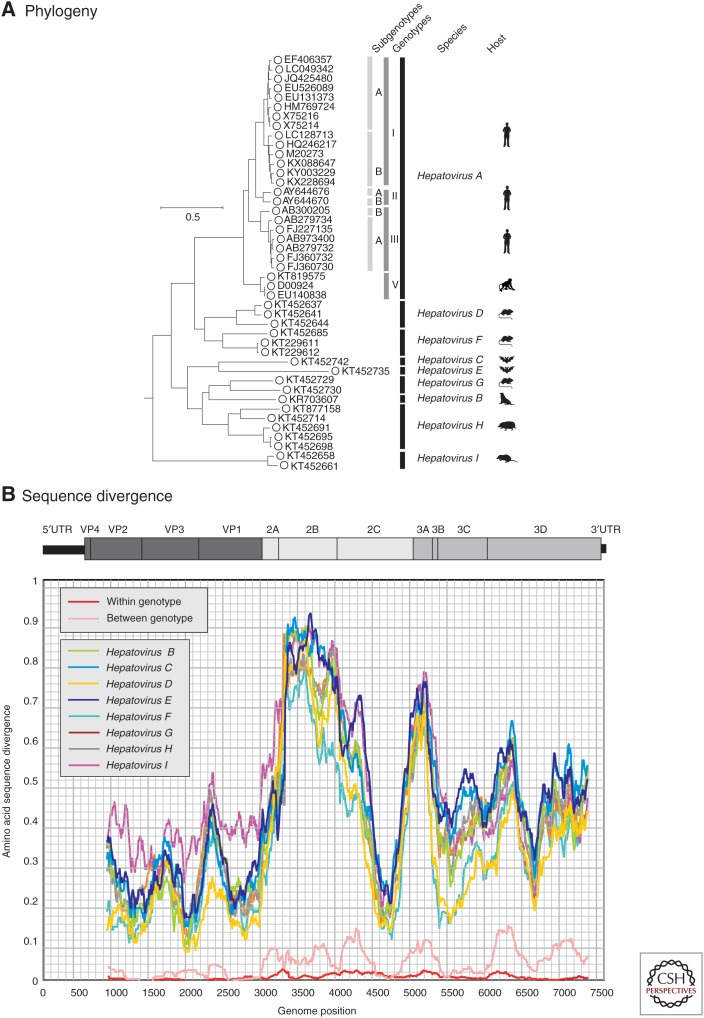
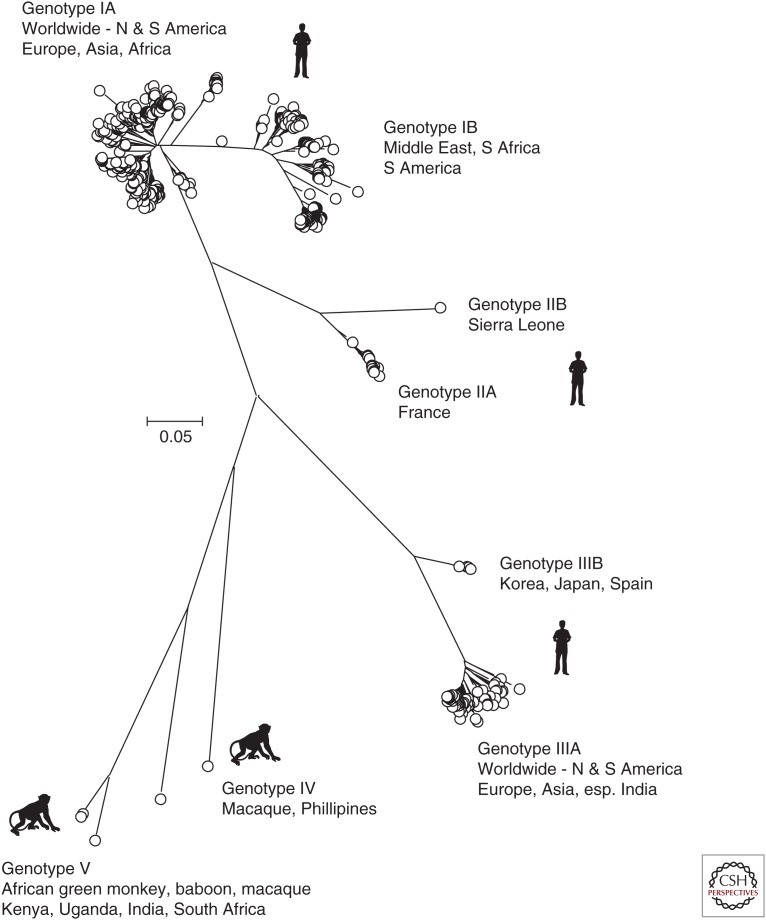
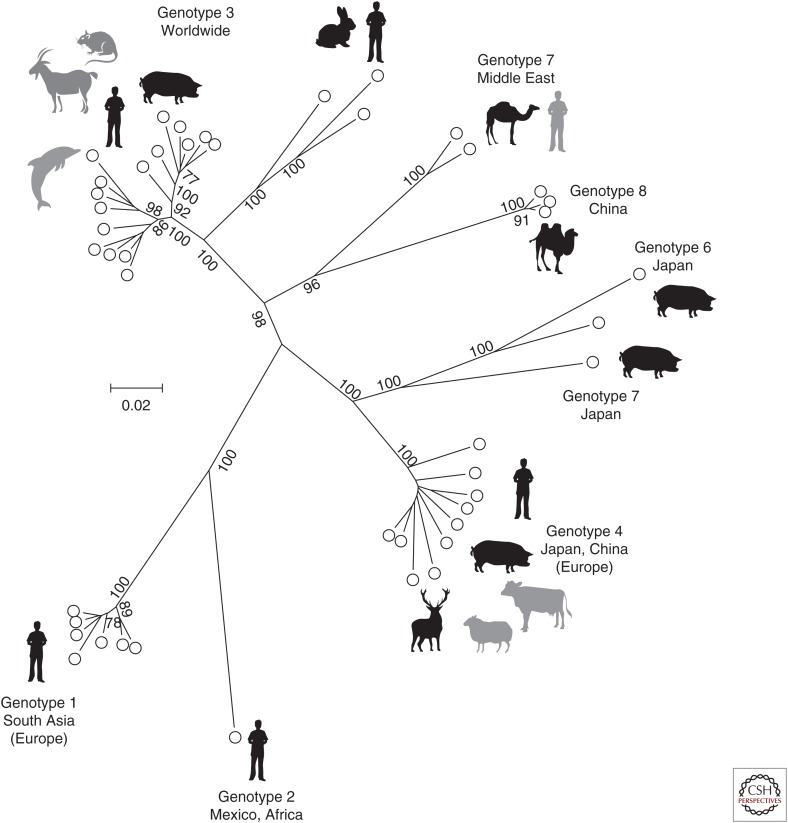
Hepatitis A virus (HAV) and hepatitis E virus (HEV) are significant human pathogens and are responsible for a substantial proportion of cases of severe acute hepatitis worldwide. Genetically, both viruses are heterogeneous and are classified into several genotypes that differ in their geographical distribution and risk group association. There is, however, little evidence that variants of HAV or HEV differ antigenically or in their propensity to cause severe disease. Genetically more divergent but primarily hepatotropic variants of both HAV and HEV have been found in several mammalian species, those of HAV being classified into eight species within the genus Hepatovirus in the virus family Picornaviridae. HEV is classified as a member of the species Orthohepevirus A in the virus family Hepeviridae, a species that additionally contains viruses infecting pigs, rabbits, and a variety of other mammalian species. Other species (Orthohepevirus B-D) infect a wide range of other mammalian species including rodents and bats.
Copyright © 2018 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures

References
-
- Arankalle VA, Ramakrishnan J. 2009. Simian hepatitis A virus derived from a captive rhesus monkey in India is similar to the strain isolated from wild African green monkeys in Kenya. J Viral Hepat 16: 214–218. - PubMed
-
- Arankalle VA, Chobe LP, Joshi MV, Chadha MS, Kundu B, Walimbe AM. 2002. Human and swine hepatitis E viruses from Western India belong to different genotypes. J Hepatol 36: 417–425. - PubMed
-
- Batts W, Yun S, Hedrick R, Winton J. 2011. A novel member of the family Hepeviridae from cutthroat trout (Oncorhynchus clarkii). Virus Res 158: 116–123. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources