

Impact of altered phosphorylation on loss of function of juvenile Parkinsonism-associated genetic variants of the E3 ligase parkin
- PMID: 29530980
- PMCID: PMC5925814
- DOI: 10.1074/jbc.RA117.000605
Impact of altered phosphorylation on loss of function of juvenile Parkinsonism-associated genetic variants of the E3 ligase parkin
Abstract
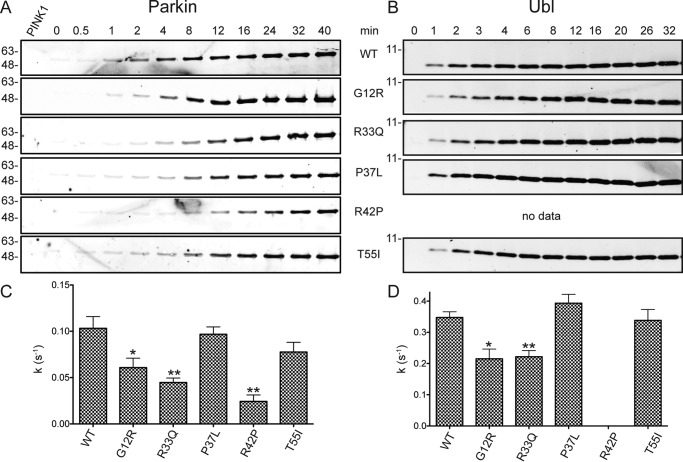
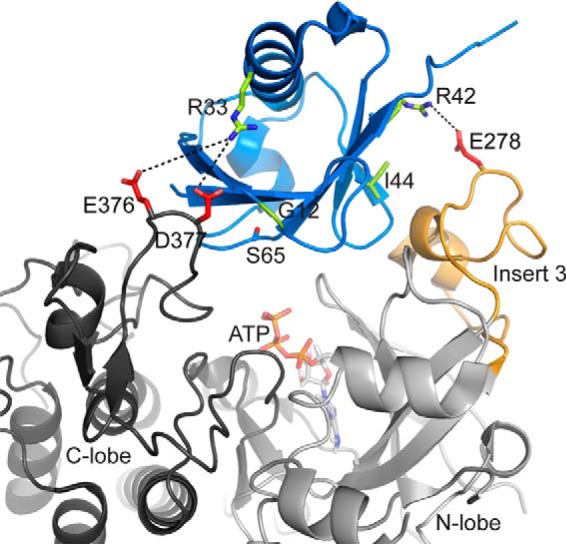
Autosomal recessive juvenile Parkinsonism (ARJP) is an inherited neurodegenerative disease in which 50% of affected individuals harbor mutations in the gene encoding the E3 ligase parkin. Parkin regulates the mitochondrial recycling pathway, which is induced by oxidative stress. In its native state, parkin is auto-inhibited by its N-terminal ubiquitin-like (Ubl) domain, which blocks the binding site for an incoming E2∼ubiquitin conjugate, needed for parkin's ubiquitination activity. Parkin is activated via phosphorylation of Ser-65 in its Ubl domain by PTEN-induced putative kinase 1 (PINK1) and a ubiquitin molecule phosphorylated at a position equivalent to Ser-65 in parkin. Here we have examined the underlying molecular mechanism of phosphorylation of parkin's Ubl domain carrying ARJP-associated substitutions and how altered phosphorylation modulates parkin activation and ubiquitination. We found that three substitutions in the Ubl domain (G12R, R33Q, and R42P) significantly decrease PINK1's ability to phosphorylate the Ubl domain. We noted that two basic loss-of-function substitutions (R33Q and R42P) are close to acidic patches in the proposed PINK1-parkin interface, indicating that ionic interactions at this site may be important for efficient parkin phosphorylation. Increased auto-ubiquitination with unique ubiquitin chain patterns was observed for two other Ubl domain substitutions (G12R and T55I), suggesting that these substitutions, along with phosphorylation, increase parkin degradation. Moreover, Ubl domain phosphorylation decreased its affinity for the potential effector protein ataxin-3, which edits ubiquitin chain building by parkin. Overall, our work provides a framework for the mechanisms of parkin's loss-of-function, indicating an interplay between ARJP-associated substitutions and phosphorylation of its Ubl domain.
Keywords: PTEN-induced putative kinase 1 (PINK1); Parkinson's disease (autosomal recessive, early onset) 7 (PARK7); parkin; protein folding; protein structure; protein–protein interaction; ubiquitylation (ubiquitination).
© 2018 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures





References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
