

Cost-effective generation of precise label-free quantitative proteomes in high-throughput by microLC and data-independent acquisition
- PMID: 29531254
- PMCID: PMC5847575
- DOI: 10.1038/s41598-018-22610-4
Cost-effective generation of precise label-free quantitative proteomes in high-throughput by microLC and data-independent acquisition
Abstract
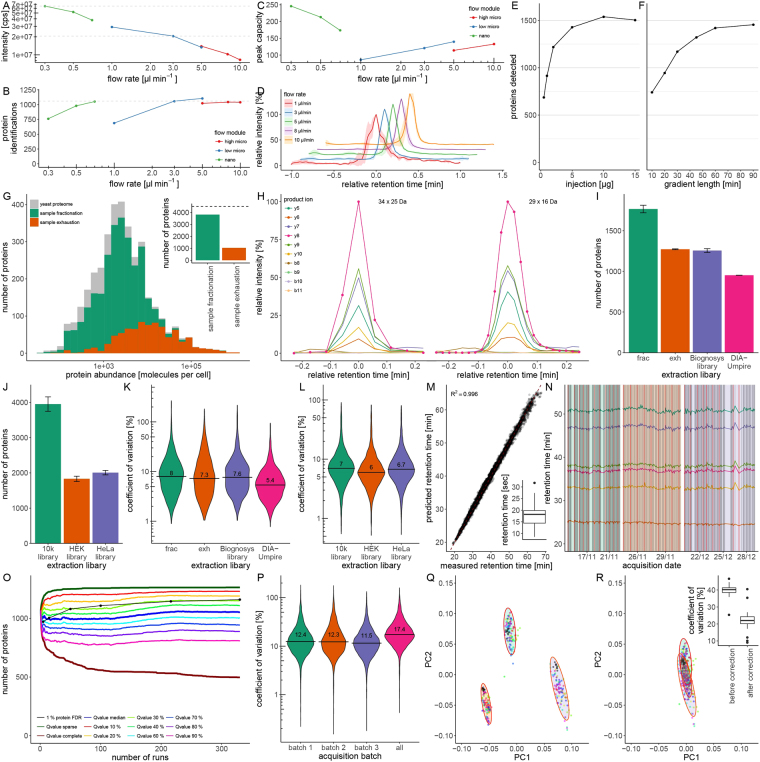
Quantitative proteomics is key for basic research, but needs improvements to satisfy an increasing demand for large sample series in diagnostics, academia and industry. A switch from nanoflowrate to microflowrate chromatography can improve throughput and reduce costs. However, concerns about undersampling and coverage have so far hampered its broad application. We used a QTOF mass spectrometer of the penultimate generation (TripleTOF5600), converted a nanoLC system into a microflow platform, and adapted a SWATH regime for large sample series by implementing retention time- and batch correction strategies. From 3 µg to 5 µg of unfractionated tryptic digests that are obtained from proteomics-typical amounts of starting material, microLC-SWATH-MS quantifies up to 4000 human or 1750 yeast proteins in an hour or less. In the acquisition of 750 yeast proteomes, retention times varied between 2% and 5%, and quantified the typical peptide with 5-8% signal variation in replicates, and below 20% in samples acquired over a five-months period. Providing precise quantities without being dependent on the latest hardware, our study demonstrates that the combination of microflow chromatography and data-independent acquisition strategies has the potential to overcome current bottlenecks in academia and industry, enabling the cost-effective generation of precise quantitative proteomes in large scale.
Conflict of interest statement
The authors J.V., R.B. and L.R. are employees of Biognosys AG (Switzerland). Spectronaut is a trademark of Biognosys AG.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
