

Microbial metagenome of urinary tract infection
- PMID: 29531289
- PMCID: PMC5847550
- DOI: 10.1038/s41598-018-22660-8
Microbial metagenome of urinary tract infection
Abstract
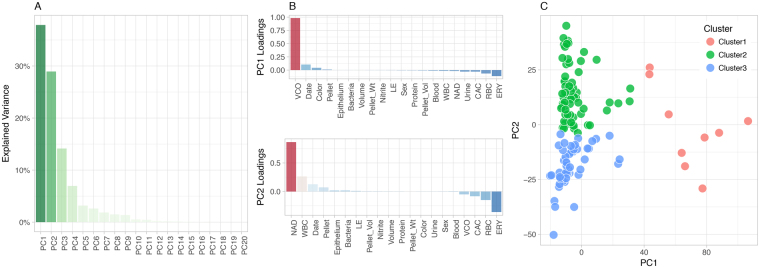
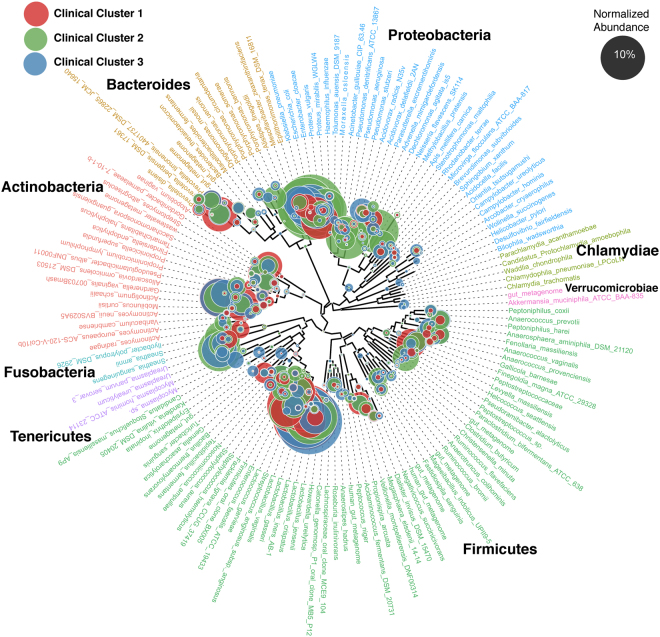
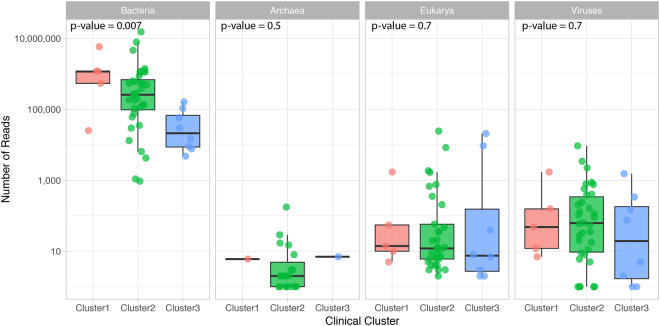
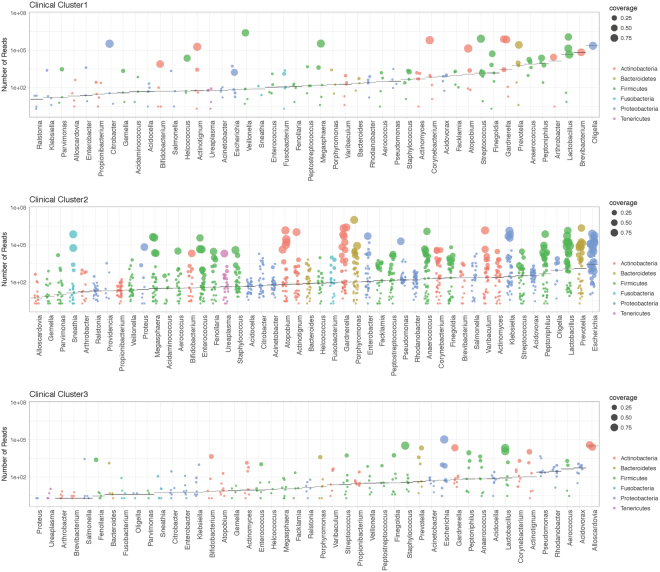
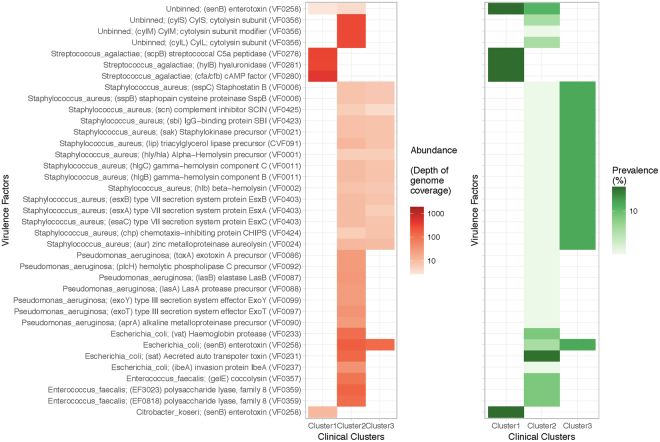
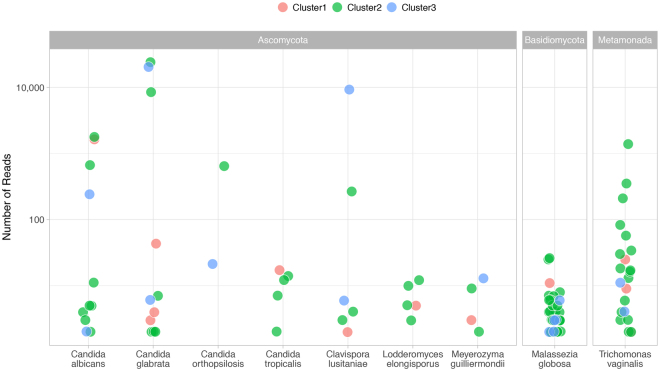
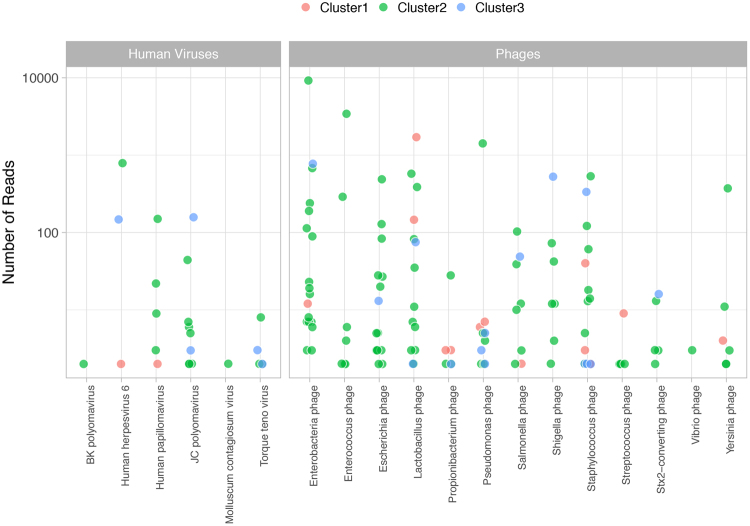
Urine culture and microscopy techniques are used to profile the bacterial species present in urinary tract infections. To gain insight into the urinary flora, we analyzed clinical laboratory features and the microbial metagenome of 121 clean-catch urine samples. 16S rDNA gene signatures were successfully obtained for 116 participants, while metagenome sequencing data was successfully generated for samples from 49 participants. Although 16S rDNA sequencing was more sensitive, metagenome sequencing allowed for a more comprehensive and unbiased representation of the microbial flora, including eukarya and viral pathogens, and of bacterial virulence factors. Urine samples positive by metagenome sequencing contained a plethora of bacterial (median 41 genera/sample), eukarya (median 2 species/sample) and viral sequences (median 3 viruses/sample). Genomic analyses suggested cases of infection with potential pathogens that are often missed during routine urine culture due to species specific growth requirements. While conventional microbiological methods are inadequate to identify a large diversity of microbial species that are present in urine, genomic approaches appear to more comprehensively and quantitatively describe the urinary microbiome.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
