

Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules
- PMID: 29532027
- PMCID: PMC5833007
- DOI: 10.1021/acscentsci.7b00572
Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules
Abstract
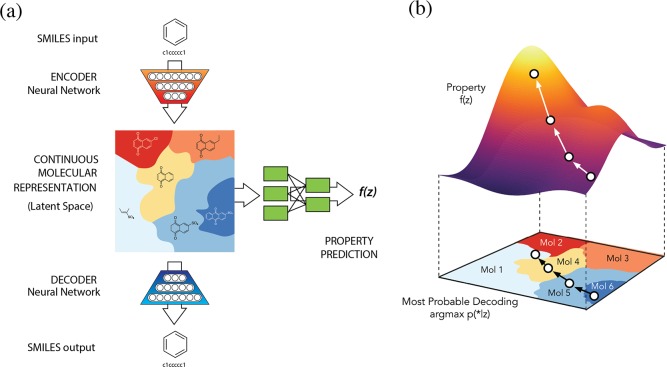
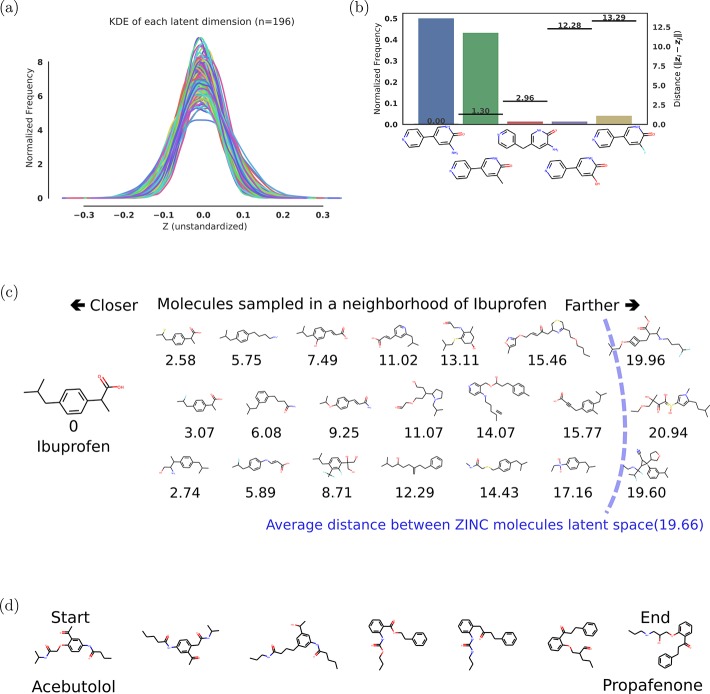
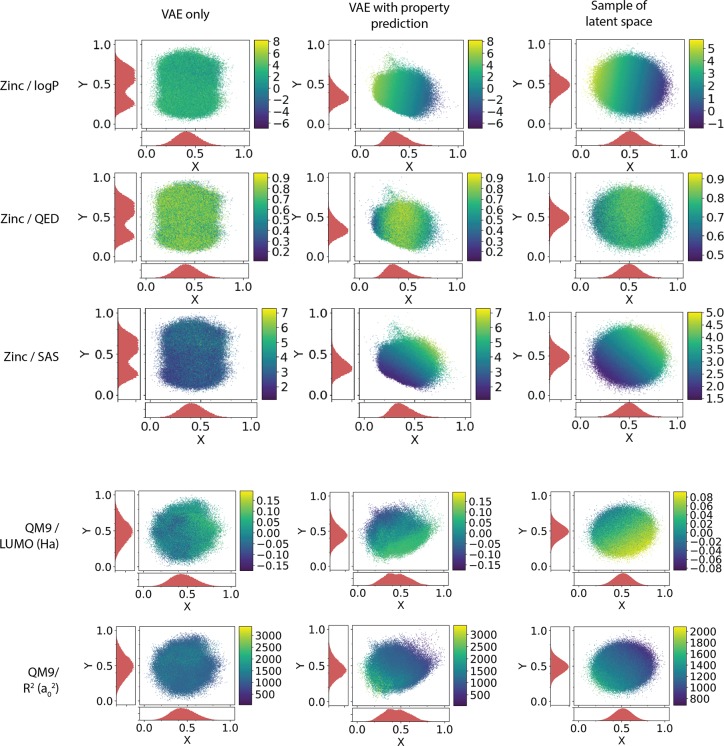
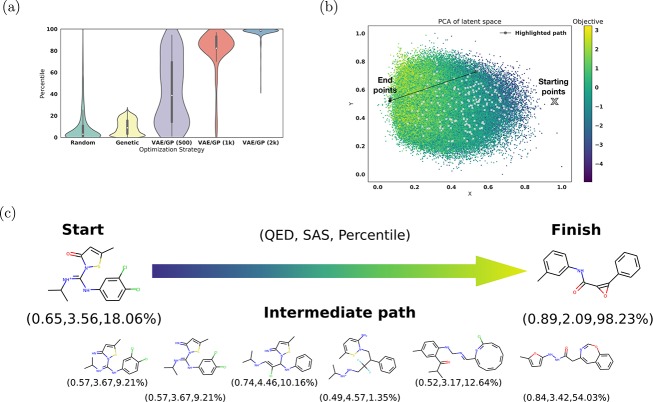
We report a method to convert discrete representations of molecules to and from a multidimensional continuous representation. This model allows us to generate new molecules for efficient exploration and optimization through open-ended spaces of chemical compounds. A deep neural network was trained on hundreds of thousands of existing chemical structures to construct three coupled functions: an encoder, a decoder, and a predictor. The encoder converts the discrete representation of a molecule into a real-valued continuous vector, and the decoder converts these continuous vectors back to discrete molecular representations. The predictor estimates chemical properties from the latent continuous vector representation of the molecule. Continuous representations of molecules allow us to automatically generate novel chemical structures by performing simple operations in the latent space, such as decoding random vectors, perturbing known chemical structures, or interpolating between molecules. Continuous representations also allow the use of powerful gradient-based optimization to efficiently guide the search for optimized functional compounds. We demonstrate our method in the domain of drug-like molecules and also in a set of molecules with fewer that nine heavy atoms.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
