

Testing of Alignment Parameters for Ancient Samples: Evaluating and Optimizing Mapping Parameters for Ancient Samples Using the TAPAS Tool
- PMID: 29533977
- PMCID: PMC5867878
- DOI: 10.3390/genes9030157
Testing of Alignment Parameters for Ancient Samples: Evaluating and Optimizing Mapping Parameters for Ancient Samples Using the TAPAS Tool
Abstract
High-throughput sequence data retrieved from ancient or other degraded samples has led to unprecedented insights into the evolutionary history of many species, but the analysis of such sequences also poses specific computational challenges. The most commonly used approach involves mapping sequence reads to a reference genome. However, this process becomes increasingly challenging with an elevated genetic distance between target and reference or with the presence of contaminant sequences with high sequence similarity to the target species. The evaluation and testing of mapping efficiency and stringency are thus paramount for the reliable identification and analysis of ancient sequences. In this paper, we present 'TAPAS', (Testing of Alignment Parameters for Ancient Samples), a computational tool that enables the systematic testing of mapping tools for ancient data by simulating sequence data reflecting the properties of an ancient dataset and performing test runs using the mapping software and parameter settings of interest. We showcase TAPAS by using it to assess and improve mapping strategy for a degraded sample from a banded linsang (Prionodon linsang), for which no closely related reference is currently available. This enables a 1.8-fold increase of the number of mapped reads without sacrificing mapping specificity. The increase of mapped reads effectively reduces the need for additional sequencing, thus making more economical use of time, resources, and sample material.
Keywords: alignment sensitivity/specificity; ancient DNA; palaeogenomics; paleogenomics; short-read mapping.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
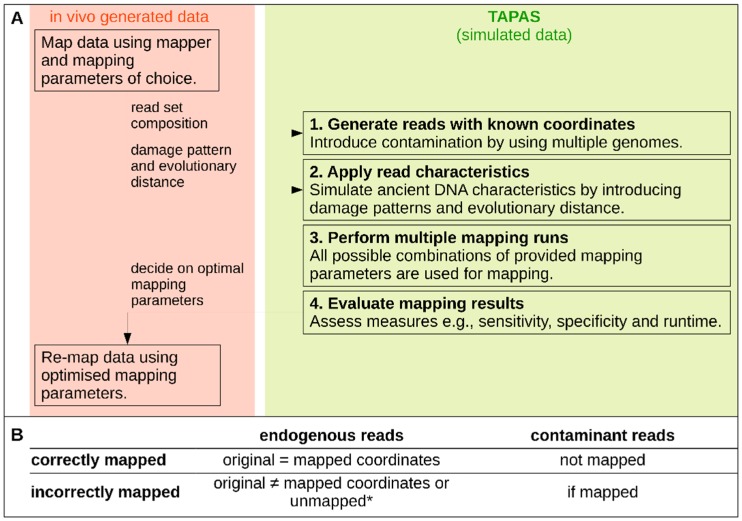
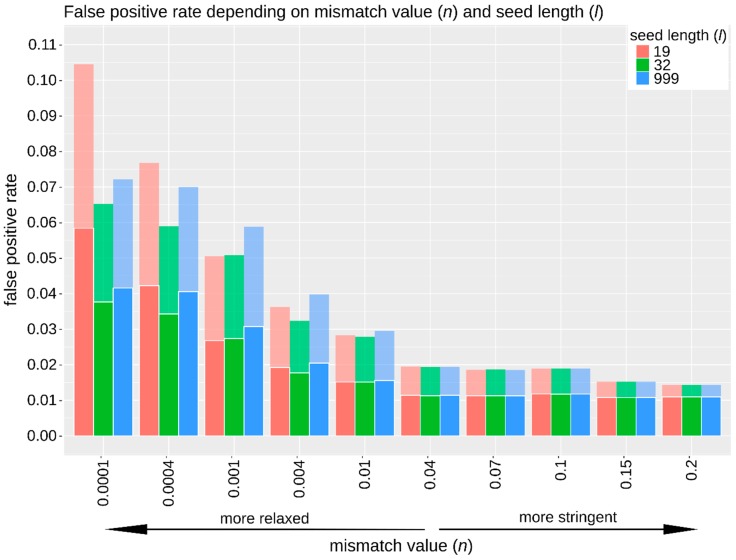
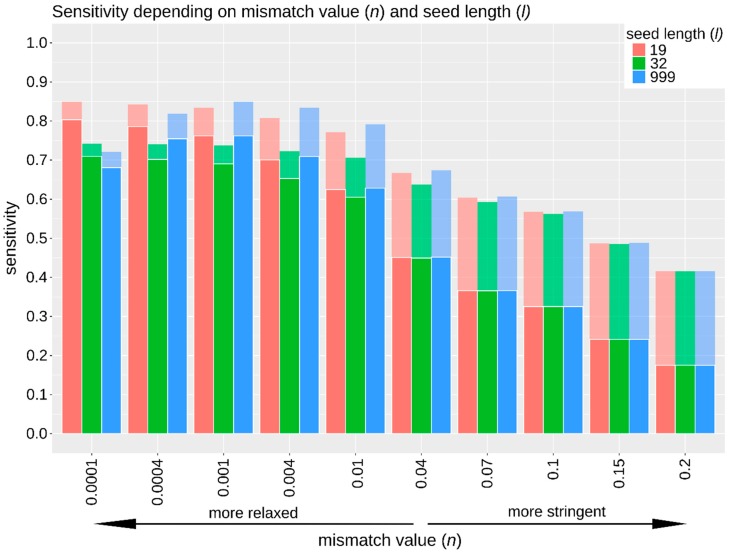
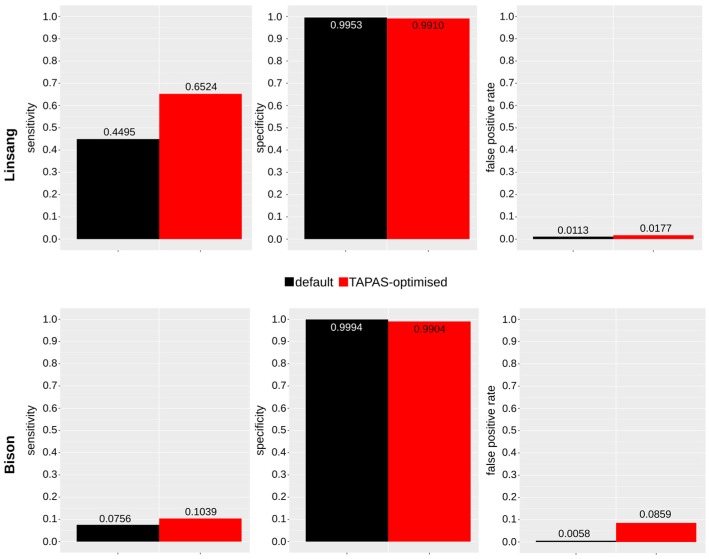
References
-
- Schubert M., Ermini L., Sarkissian C.D., Jónsson H., Ginolhac A., Schaefer R., Martin M.D., Fernández R., Kircher M., McCue M., et al. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 2014;9:1056–1082. doi: 10.1038/nprot.2014.063. - DOI - PubMed
-
- Green R.E., Malaspinas A.-S., Krause J., Briggs A.W., Johnson P.L.F., Uhler C., Meyer M., Good J.M., Maricic T., Stenzel U., et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell. 2008;134:416–426. doi: 10.1016/j.cell.2008.06.021. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
