

Liddle Syndrome: Review of the Literature and Description of a New Case
- PMID: 29534496
- PMCID: PMC5877673
- DOI: 10.3390/ijms19030812
Liddle Syndrome: Review of the Literature and Description of a New Case
Abstract
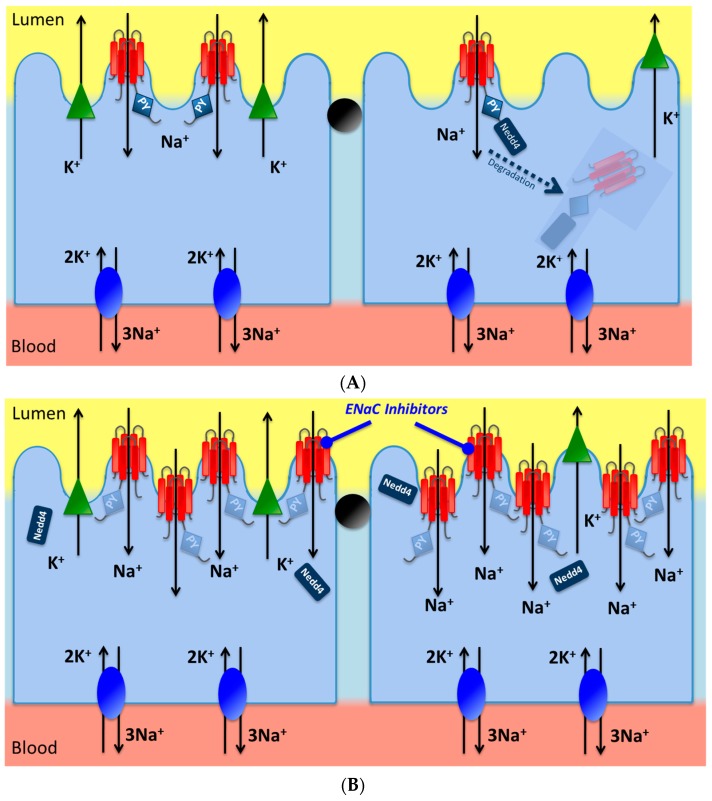
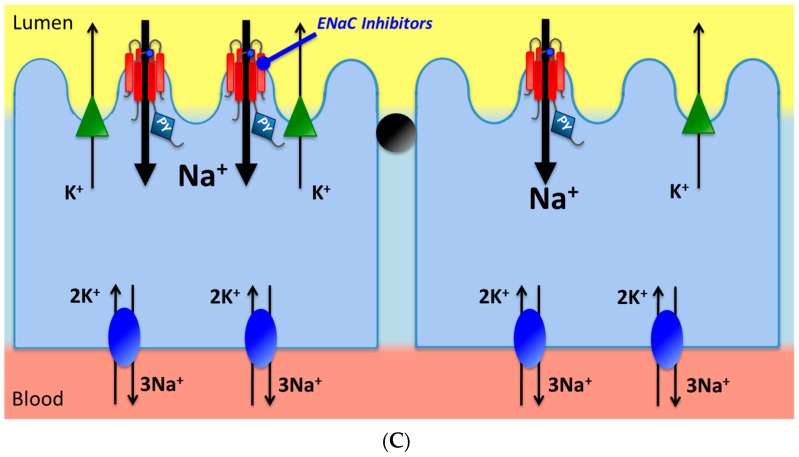
Liddle syndrome is an inherited form of low-renin hypertension, transmitted with an autosomal dominant pattern. The molecular basis of Liddle syndrome resides in germline mutations of the SCNN1A, SCNN1B and SCNN1G genes, encoding the α, β, and γ-subunits of the epithelial Na⁺ channel (ENaC), respectively. To date, 31 different causative mutations have been reported in 72 families from four continents. The majority of the substitutions cause an increased expression of the channel at the distal nephron apical membrane, with subsequent enhanced renal sodium reabsorption. The most common clinical presentation of the disease is early onset hypertension, hypokalemia, metabolic alkalosis, suppressed plasma renin activity and low plasma aldosterone. Consequently, treatment of Liddle syndrome is based on the administration of ENaC blockers, amiloride and triamterene. Herein, we discuss the genetic basis, clinical presentation, diagnosis and treatment of Liddle syndrome. Finally, we report a new case in an Italian family, caused by a SCNN1B p.Pro618Leu substitution.
Keywords: Liddle syndrome; SCNN1A; SCNN1B; SCNN1G; hypertension; hypokalemia; low renin hypertension; monogenic hypertension.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Olsen M.H., Angell S.Y., Asma S., Boutouyrie P., Burger D., Chirinos J.A., Damasceno A., Delles C., Gimenez-Roqueplo A.P., Hering D., et al. A call to action and a lifecourse strategy to address the global burden of raised blood pressure on current and future generations: The Lancet Commission on hypertension. Lancet. 2016;388:2665–2712. doi: 10.1016/S0140-6736(16)31134-5. - DOI - PubMed
-
- Liddle G.W., Bledsoe T., Coppage W.S.J. A familial renal disorder simulating primary aldosteronism but with negligible aldosterone secretion. Trans. Assoc. Am. Phys. 1963;76:199–213.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
